INTRODUCTION
Attention-deficit/hyperactivity disorder (ADHD) is a common neurobehavioral disorder, affecting approximately 5% of school-age children worldwide,1 and an estimated 5.9% of children in Korean elementary schools.2
Treatment for ADHD aims to reduce symptoms, enhance functionality and improve individual and family well-being.3 Standardized stimulant pharmacotherapy for ADHD is associated with superior symptom reduction compared with psychotherapy alone but up to 30% of patients may not respond or are intolerant to stimulant medications.4 Atomoxetine is a novel non-stimulant treatment for ADHD with a different mode of action to other currently approved (stimulant) medications.5 The efficacy and safety of atomoxetine in treating ADHD has been demonstrated in numerous randomized, double-blind, placebo-controlled trials and open-label studies in children, adolescents and adult patients.4 These studies have included over 4,000 child and adolescent patients, of whom over 1,100 have been followed for more than 6 months, establishing the efficacy and tolerability of atomoxetine in the treatment of ADHD.4,6-14
A dose-response relationship for atomoxetine was demonstrated in a pivotal fixed-dose, double-blind, randomized, controlled trial in the United States, in which atomoxetine 1.2 mg/kg/day was identified as an optimal dose for most patients.11 Although the majority of studies investigating atomoxetine have been conducted in predominantly Caucasian populations, it has also been shown to be effective and well-tolerated by children and adolescents in Asia, including in Taiwan, China, Japan and Korea.15-17 However, the dose response of atomoxetine in these populations has not been extensively studied.
Atomoxetine has received approval for the treatment of children and adolescents with ADHD from the Korean Food and Drug Administration but the dose response of atomoxetine in Korean pediatric patients has not been studied specifically. The primary aim of this study, therefore, was to assess the response to three different doses of atomoxetine in Korean pediatric outpatients with ADHD. The safety and tolerability of atomoxetine at each dose were also assessed.
METHODS
Partipants
All patients enrolled in this study conducted in Korea were children or adolescents of Korean origin aged 6 to 18 years, with a diagnosis of ADHD as defined by The American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) diagnostic criteria.18 Patients were eligible for enrollment if they recorded symptom severity thresholds of 1.5 standard deviations above age and gender norms (based on a large, representative sample of the North American population) on the ADHD Rating Scale-IV-Parent: Investigator-Administered and Scored (ADHDRS-IV-Parent:Inv),19 and a Clinical Global Impressions-ADHD-Severity (CGI-S) score ≥4 at Visits 1 and 2. Enrolled patients also had to meet all of the following criteria: did not take any medication for ADHD treatment at least 2 weeks prior to randomization and at least 1 week prior to obtaining baseline ADHDRS-IV-Parent:Inv and CGI-S scores; had no significant laboratory abnormalities or clinical conditions that would preclude participation at study entry; had no impairment in intelligence as assessed clinically by the investigator; and were able (along with parents or legal guardian) to keep appointments for clinic visits and all examinations as required by the protocol. Informed consent was obtained from all parents or legal representative, and patients where appropriate, prior to study initiation.
The study protocol was approved by the Ethical Review Board of each participant investigational hospital: Seoul National University Hospital, Soonchunhyang University Bucheon Hospital, Asan Medical Center, Yonsei University Severance Hospital, Hanyang University Medical Center, Kangbuk Samsung Hospital, and Gachon University Gil Hospital in South Korea.
Patients were not eligible for enrollment if they had participated in a previous study of atomoxetine; had a history of bipolar disorder, psychosis, autism, Asperger's syndrome, pervasive developmental disorder, or conduct disorder; had a history of any seizure disorder, or were taking anticonvulsants for seizure control; were at serious risk of suicide; were likely to need psychotropic medications other than the study drug, including Chinese medicine, herbal medicine, and/or health food supplements that had central nervous system activity. Patients were discontinued from the study if they were significantly non-compliant with the study drug (defined below), or required treatment with another therapeutic agent for the treatment of ADHD or a psychoactive medication excluded by the study protocol.
Study design
This was a Phase 3b multicenter, randomized, open-label, parallel trial with two study periods conducted to provide additional efficacy and safety information on the response to three different doses of atomoxetine within the Korean pediatric ADHD population. Enrolled patients were randomized to one of three treatment arms, with treatment given orally in two divided doses, for 6 weeks. In this open-label setting, a sub-therapeutic pseudo-placebo arm (Arm 1; atomoxetine 0.2 mg/kg/day) was utilized instead of placebo for practical and ethical reasons. The actual doses of study treatment received by patients may have varied because of the limited capsule sizes available and variations in patients' body weight. Patients in Arm 2 received atomoxetine at a target dose of 0.5 mg/kg/day and patients in Arm 3 were exposed to atomoxetine in a dose escalation fashion from a target of 0.5 mg/kg/day for 7 days followed by 0.8 mg/kg/day for 7 days, then 1.2 mg/kg/day thereafter; the dose is referred to as 1.2 mg/kg/day in this report for simplicity. The 6-week treatment period was considered appropriate based on data from previous studies of atomoxetine.
Enrolled patients were randomized by a computer generated random sequence in a 1 : 1 : 1 ratio to the three treatment arms at Visit 2. Patients were considered significantly non-compliant if they missed three consecutive doses of the study medication or failed to take at least 80% of the prescribed doses of medication during two or more visit intervals, or if they had intentionally or repeatedly taken more than 120% of the prescribed dose. All medications other than the study drug taken during the study were recorded. Patients were permitted to take over the counter medication or prescription medications not specifically excluded from the study protocol (including aspirin, cold preparations, iron supplements and vitamins). This study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki.
Efficacy and safety measures
The primary objective of the study was to assess the change in ADHD symptoms using three different doses of atomoxetine in the Korean pediatric ADHD population. The primary efficacy measure used in this study was the ADHDRS-IV-Parent: Inv total scale score,19,20 an 18-item scale corresponding with each of the 18 symptoms described by the DSM-IV-TR, which has been validated in the Korean language.21 Each item is scored on a 0- to 3-point scale, assessing symptom severity in the past week. The total score was computed as the sum of the scores for each item, and primary efficacy measured as change from baseline to last observation carried forward (LOCF) endpoint. Inattention and hyperactivity/impulsivity subscores were also computed. Secondary objectives of this study were to assess the efficacy of each dose in reducing the severity ADHD symptoms, and the safety and tolerability of atomoxetine at each dose. Secondary efficacy measures were the change from baseline to endpoint in the CGI-S rating scale and the Clinical Global Impressions-ADHD-Improvement (CGI-I) score at the end of study (6 weeks). Safety assessments included: adverse events (AEs); laboratory data (chemistry, hematology, urinalysis, urine drug screen, thyroid function test, pregnancy test if necessary), completed at Visit 1 for screening purposes; vital signs [systolic and diastolic Blood pressure (BP), body temperature, pulse rate, weight and height, and body mass index], recorded at regular intervals; Columbia Suicide-Severity Rating Scale and the "Self-Harm Supplement" Form, which were completed if an investigator suspected that an adverse event represented suicidal thought or behavior, or if a self-harm event was reported.
Statistical analyses
Efficacy analyses were performed on an intention-to-treat (ITT) basis in which all randomized patients with a baseline and at least one post-baseline ADHDRS-IV-Parent:Inv total score and CGI-S score were included and subsequently analyzed according to their original randomly assigned treatment group. Adverse event analyses were conducted on all data from all patients receiving at least one dose of the study drug and grouped by the actual maximum dose the patient received over all visits (not the assigned dose as per ITT). Patients were thus allocated to one of three groups based on the actual dose received (referred to as Dosage Groups): 1) 0.01-0.35 mg/kg/day, 2) 0.36-0.85 mg/kg/day, 3) >0.85 mg/kg/day. These groups overlapped almost entirely with the treatment arms based on target doses used in the ITT analyses. The percentages of randomized patients who completed or discontinued Study Period II were summarized overall and by treatment arm, and possible treatment differences were assessed using Fisher's exact test. Patient discontinuation in Study Period I was summarized by percentages and counts. Patient baseline characteristics (demographics, ADHD characteristics, historical illness and pre-existing conditions) were summarized with descriptive statistics. The percentage of patients with significant non-compliance, and use of concomitant medication was summarized by visit and overall, and by treatment arm. Unless otherwise specified, all formal tests were conducted at a two-sided alpha level of 0.05.
Primary efficacy analysis
The primary summary statistic used to assess dose response was the least squares mean overall change from baseline to endpoint [6 weeks (LOCF)] in ADHDRS-IV-Parent:Inv total score for each treatment arm, calculated (with 95% CI) using an analysis of covariance (ANCOVA) model with a LOCF approach to impute missing data. This model included pre-specified covariates of baseline ADHDRS-IV-Parent:Inv total score, treatment arm and investigator. The primary efficacy analysis was also repeated using a longitudinal/mixed effects model with repeated measures (MMRM) approach in a sensitivity framework.
Secondary efficacy analyses
The secondary summary statistics were the change from baseline to endpoint in CGI-S and endpoint measure of CGI-I for each treatment arm. Longitudinal profiles of change from baseline score were generated using a MMRM approach for CGI-S and CGI-I data with pre-specified fixed effect covariates of baseline CGI-S score, treatment arm, visit, investigator and treatment arm-by-visit interaction, with an unstructured covariance matrix. Contrasts between treatment arms were made at Visit 5. The longitudinal analyses of CGI-S and CGI-I scores using MMRM were repeated using ANCOVA and LOCF approaches, which were compared with the protocol-specified secondary analyses as a sensitivity framework. Contrasts between treatment arms were made at 42 days, and were also compared with the protocol-specified secondary analyses as a sensitivity framework.
Additional efficacy analyses
To further support the specified primary and secondary assessments of the dose response, monotonic dose response (strictly increasing response with increasing dose) was statistically tested using a 1-sided Williams test with a 0.025 significance level.22
Determination of sample size
A sample size of 150 patients, allocated in a 1 : 1 : 1 ratio to the three treatment arms, was chosen on a pragmatic basis that this would be a sufficient number of patients to adequately provide a meaningful dose response plot as well as useful safety and tolerability profiles.
RESULTS
Patient disposition
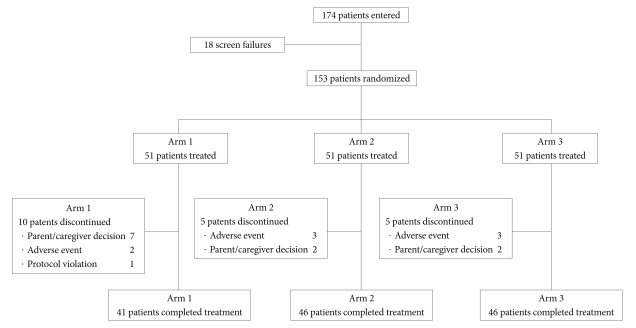
The study cohort disposition is summarized in Figure 1. Safety analyses were conducted on the as-treated population (n=153) according to the actual atomoxetine dose taken during the study. Consequently, the safety analysis assessed 51 patients in Dosage Group 1, 54 patients in Dosage Group 2, and 48 patients in Dosage Group 3. The majority of patients in all three arms were compliant with treatment overall [2 patients (3.9%) in Arms 1 and 3, and 3 patients in Arm 2 (5.9%) were deemed significantly non-compliant]. The most common previous treatment class (25.5% overall: Arm 1=23.5%, Arm 2=27.5%, Arm 3=25.5%) was centrally acting sympathomimetics (methylphenidate formulations). The most common post-randomization concomitant medication class (3.9% overall: Arm 1=3.9%, Arm 2=5.9%, Arm 3=2.0%) was sympathomimetics (pseudoephedrine and phenylephrine).
Baseline demographics
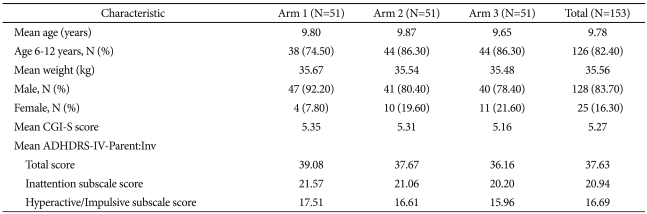
Table 1 summarizes the baseline demographics and disease characteristics for all randomized patients.
Primary efficacy analysis: ADHDRS-IV-Parent:Inv
Figure 2 provides a graphical representation of the primary efficacy analysis results, demonstrating the evident monotonic ordering of dose response. The least-squares mean changes from baseline to endpoint ADHDRS-IV-Parent:Inv total scores were -9.6, -12.3, and -14.5 with atomoxetine 0.2, 0.5 and 1.2 mg/kg/day, respectively (p=0.024 - F-test).
The ANCOVA model for ADHDRS-IV-Parent:Inv total score demonstrated a significantly greater improvement in mean change from baseline to endpoint for atomoxetine 1.2 mg/kg/day in a pairwise comparison with atomoxetine 0.2 mg/kg/day (p=0.006), and the mean difference of 4.96 after 6 weeks between these treatment arms was considered clinically significant. Pairwise comparisons for mean change from baseline to endpoint ADHDRS-IV-Parent: Inv score showed a mean difference of 2.21 (p=0.20) between atomoxetine 1.2 and 0.5 mg/kg/day, and 2.76 (p=0.12) for 0.5 and 0.2 mg/kg/day.
Secondary efficacy analyses-mean change from baseline CGI-S score
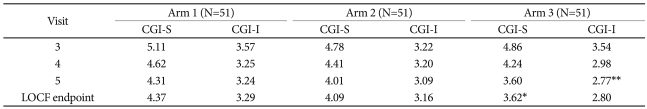
Overall, patients were "moderately" ill at endpoint (mean CGI-S score: 4.02), compared with "markedly" ill at baseline (mean CGI-S score: 5.27). Table 2 illustrates the mean CGI-S score by visit for all treatment arms. Atomoxetine 1.2 mg/kg/day was associated with greater improvement from baseline to endpoint CGI-S score compared with atomoxetine 0.2 mg/kg/day at 6 weeks (-1.54 versus -0.98; p=0.011).
Secondary efficacy analyses-mean CGI-I score
Table 2 illustrates the mean CGI-I score by visit for all treatment arms. Patients receiving atomoxetine 1.2 mg/kg/day showed greater improvement in mean CGI-I score than patients receiving atomoxetine 0.2 mg/kg/day at 6 weeks (mean CGI-I score: 2.80 versus 3.29; p=0.0025).
Additional analyses
The null hypothesis addressed by the Williams test maintains that the mean response for all dose groups is equal while the alternative hypothesis maintains that a monotone dose-response relationship exists. The result comparing the 1.2 and 0.2 mg/kg/day treatment arms (p=0.016) suggests that there is strong evidence to reject the null hypothesis in favor of a monotone dose-response relationship.
Safety and tolerability
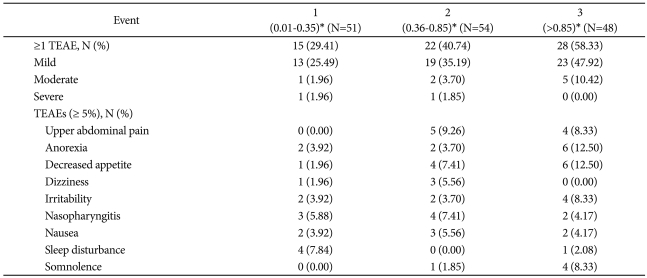
There were no deaths reported in this study. Two patients [one patient in each of Dosage Group 2 (0.36-0.85 mg/kg/day) and 3 (>0.85 mg/kg/day)] experienced serious AEs that were considered by the investigator to be unrelated to the study drug. Eight patients discontinued due to AEs: two patients in Arm 1, three patients in Arm 2, and three patients in Arm 3 (Figure 1).
The most frequently reported AEs (occurring in ≥5% of patients) are shown in Table 3. The majority of these events were mild or moderate, and no events related to suicide ideation or self-harm were reported. More patients receiving atomoxetine at approximately 1.2 mg/kg/day (Dosage Group 3) reported ≥1 treatment-emergent adverse event/s (58.3%) compared with 0.5 (Dosage Group 2; 40.7%; p=0.11) or 0.2 mg/kg/day (Dosage Group 1; 29.4%; p=0.005).
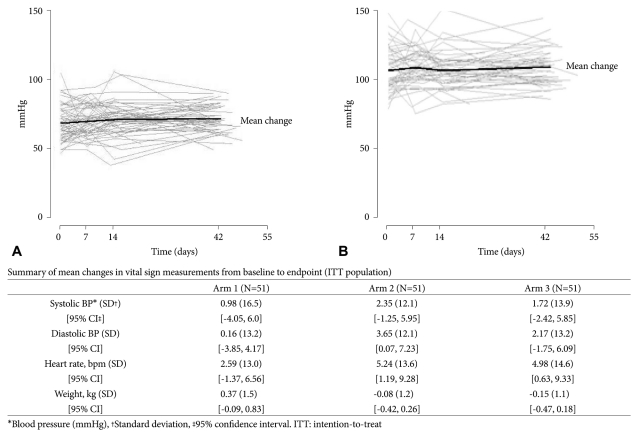
BP fluctuations were observed for some patients at the beginning of the study, although these were not unidirectional and tended to normalize during the study period. No clinically significant mean changes in systolic or diastolic BP from baseline to endpoint were observed between treatment groups. Longitudinal changes in the systolic and diastolic BP of patients in Arm 3 (1.2 mg/kg/day target dose), and a summary of the mean change in vital signs measurements for all treatment arms from baseline to endpoint are shown in Figure 3.
DISCUSSION
This is the first study evaluating the dose response of atomoxetine for treating ADHD in a Korean population. A monotonic ordering of dose response for atomoxetine prescribed at 0.2 mg/kg/day, 0.5 mg/kg/day, and 1.2 mg/kg/day in the improvement of ADHD symptoms in Korean children and adolescents following 6 weeks of treatment was demonstrated. The additional sensitivity analyses provided strong supportive evidence of the monotonic dose-response relationship observed in the primary efficacy analysis. Furthermore, atomoxetine 1.2 mg/kg/day showed clinically meaningful and statistically significant improvements in ADHD symptoms compared with atomoxetine 0.2 mg/kg/day. Most treatment-emergent AEs observed with all three doses of atomoxetine were of mild or moderate severity and no unexpected safety findings were reported.
The monotonic ordering of dose response observed here strongly reflects observations in other studies conducted worldwide. In the pivotal fixed-dose, 8-week, placebo-controlled study of atomoxetine in the treatment of ADHD in children and adolescents in the United States, atomoxetine 1.2 mg/kg/day demonstrated a mean change from baseline to endpoint ADHDRS-IV-Parent:Inv total score of -13.6 (SD 14.0).11,23 In this study, atomoxetine 1.2 mg/kg/day produced a similar mean change from baseline to endpoint ADHDRS-IV-Parent: Inv total score of -14.5 (SD 9.71), providing evidence that atomoxetine 1.2 mg/kg/day is as effective in Korean pediatric and adolescent patients as it is in patients from the United States. These data also suggest that atomoxetine 1.2 mg/kg/day and 0.5 mg/kg/day provide clinically superior efficacy compared with 0.2 mg/kg/day in the Korean population. However, the study was not principally designed to make pairwise comparisons between treatment arms and it is therefore difficult to draw further conclusions from these data. The data presented in this study are consistent with observations of an atomoxetine dose response in Caucasian pediatric ADHD populations and also support the limited data available from studies in Korea, China, Taiwan and Japan showing the efficacy of atomoxetine in Asian pediatric ADHD patients.15-17
All three doses of atomoxetine evaluated in this study proved to be safe and well-tolerated by Korean pediatric patients. Adverse event rates were generally dose-dependent; atomoxetine >0.85 mg/kg/day was associated with higher rates of AEs than 0.36-0.85 mg and 0.01-0.35 mg/kg/day, but most treatment-related AEs were mild to moderate and are consistent with those observed in global studies.4,7,11,13,23-25 While it has been suggested that the Korean population may experience AEs at relatively lower doses of psychotropic medications,26 the incidence of AEs reported here are lower than those reported in other studies of atomoxetine (0.8-1.8 mg/kg/day) in Asian and Hispanic pediatric populations.15,17 Importantly, the high completion rate of 86.9% observed in this study emphasizes the favorable tolerability of atomoxetine in Korean pediatric patients. BP fluctuations were observed early in this study, although BP normalized within the study period in most patients and there was no significant increase in BP reported between treatment arms. This is consistent with findings in studies of Caucasian and non-Caucasian pediatric patients7,11-13,15,17,24,26 and may be expected given the increases in noradrenergic tone observed with atomoxetine.27
The interpretation of these data is limited by several factors. Most importantly, the study was not designed principally to make pairwise comparisons between treatment arms. It is also possible that the open-label study design may have introduced investigator or patient and parent bias in favor of all doses, which could have influenced outcomes. Additionally, the study assessed only core ADHD symptoms using the ADHDRS-IV-Parent: Inv, CGI-S and CGI-I scales; the data do not describe the severity or prevalence of ADHD comorbidities in this population, which may have also influenced study assessments. Furthermore, the relatively short 6-week study period means that long term safety issues cannot be addressed.
Overall, atomoxetine was found to be safe and well tolerated at all doses administered in the Korean patients participating in the study, and 1.2 mg/kg/day was found to be an efficacious dose in this patient population.