Bright Light Therapy in the Morning or at Mid-Day in the Treatment of Non-Seasonal Bipolar Depressive Episodes (LuBi): Study Protocol for a Dose Research Phase I / II Trial
Article information
Abstract
Objective
This study protocol aims to determine, using a rigorous approach in patients with bipolar disorder (BD) and non-seasonal major depressive episode (MDE), the characteristics of bright light therapy (BLT) administration (duration, escalation, morning and mid-day exposures) depending on the tolerance (hypomanic symptoms).
Methods
Patients with BD I or II and treated by a mood stabilizer are eligible. After 1 week of placebo, patients are randomized between either morning or mid-day exposure for 10 weeks of active BLT with glasses using a dose escalation at 7.5, 10, 15, 30 and 45 minutes/day. A further follow-up visit is planned 6 months after inclusion. Patients will be included by cohorts of 3, with at least 3 days of delay between them, and 1 week between cohorts. If none meet a dose limiting toxicity (DLT; i.e hypomanic symptoms), the initiation dose of the next cohort will be increased. If one patient meet a DLT, an additionnal cohort will start at the same dose. If 2 or 3 patients meet a DLT, from the same cohort or from two cohorts at the same dose initiation, the maximum tolerated dose is defined. This dose escalation will also take into account DLTs observed during the intra-subject escalation on previous cohorts, with a “Target Ceiling Dose” defined if 2 DLTs occured at a dose.
Discussion
Using an innovative and more ergonomic device in the form of glasses, this study aims to better codify the use of BLT in BD to ensure a good initiation and tolerance.
Trial registration
ClinicalTrials.gov Identifier: NCT03396744.
BACKGROUND
Bipolar disorder (BD) is a severe brain disorder characterized by the recurrence of mood episodes. About 1% to 4% of the worldwide population suffers from BD, ranking as the fourth most important contributor to the global disease burden among mental, neurological and substance-use disorders [1,2]. BD is associated with a 10–20 years decrease in life expectancy, which has not improved over recent decades, unlike the pattern seen in the general population [3,4]. This severity of BD is closely associated with a high relapse rate, with 60–80% of relapses occurring within 2 years after a manic or depressive episode [5-7].
Patients presenting with BD spend more time with depressive symptoms than with manic ones, which have a major impact on the quality of life and is associated with poorer outcomes including recurrences and suicide. Indeed, a 13-year follow-up study of patients with BD type I demonstrated that depressive symptoms predominated over (hypo)manic symptoms (32% vs. 9% of total follow-up weeks, respectively) [8]. Similarly, patients with BD type II suffer more from depressive symptoms than (hypo)manic symptoms (50% vs. 1% of total follow-up weeks, respectively) [9]. These observations are confirmed up to 20 years of follow-up, both in BD type I and II, with a clear predominance of depressive symptoms during 59% of the follow-up versus 2% for (hypo)manic symptoms [10]. However, in bipolar depression, available antidepressant strategies show low efficacy apart from electroconvulsive therapy (ECT) [11,12]. Mood stabilizers in monotherapy rarely suffices to treat depression and conventionnal antidepressant are added but carry the risk of serious side effects, including an affective switch from depression to hypomania/mania, rapid cycling, and suicidality [11,12]. Given this rather bleak therapeutic landscape, there is an urgent need to develop new antidepressant treatments in BD.
The use of light for its antidepressant action dates back to the beginnings of civilization [13], and a growing body of evidence has been generated over the last decade about bright light therapy (BLT, also named phototherapy) evolving as an effective depression treatment not only to be used in seasonal affective disorder [14] (SAD, a syndrome defined by recurrent depressions that occur annually at the same time each year [15]), but also in non-seasonal depression both in unipolar and bipolar disorders [13]. Indeed, BLT is the cornerstone treatment of seasonal affective disorders since more than two decades [16,17] with a first case series published in 1984 by Rosenthal and colleagues [15]. Artificial bright light appeared more effective than dim light, but also showed the effects of lengthening the photoperiod (defined as the length of light period during a night/day cycle) and suppressing the melatonin secretion [15]. BLT is the first line treatment for depression with seasonal patterns with an efficacy equivalent to typical antidepressants like selective serotonin reuptake inhibitors (SSRIs) [18]. This benefit was confirmed in meta-analyses of randomized trials evaluating BLT versus placebo in subjects with seasonal unipolar depression [19], and seasonal bipolar depression [20]. Interestingly, it has been recently demonstrated that BLT may also be useful in non-seasonal depression. Indeed, BLT appeared efficacious and well tolerated in the treatment of adults with non-seasonal unipolar depression, of moderate to severe intensity, with an effect size equal or superior to SSRIs [19,21], and was confirmed in a meta-analysis [22].
Despite these new insights about light antidepressant action, the management of BLT continues to be a point of debate, especially for BD with non-seasonal pattern [13]. There is only one randomized negative study in BD that recruited 44 non-seasonal patients but whose analyzes focused on the comparison of 10 patients treated with BLT versus 11 patients treated with placebo [23]. However, 10 subjects per group is probably insufficient as sample size to test the therapeutic efficacy of BLT. For example, Lam et al. [21] could identify that 54 subjects per comparison group allowed a statistical power of 80% to detect a mean difference with placebo on the MADRS depression scale of 3.5 points or an effect size of 0.4, considered as mild to moderate therapeutic effect. In addition, an older randomized controlled trial of Kripke et al. [24], that included both BD and non-BD patients (n=25) versus placebo (n=26), observed that non-seasonal patients with BD had a greater decreased of depressive symptoms than non-BD patients, although not significant. This is confirmed by a study that focused specifically on comparing the effects of light therapy on non-seasonal bipolar versus non-bipolar depressions [25]. These authors observe, in a small sample of 17 patients, that BLT is significantly more effective on bipolar than non-bipolar non-seasonal depressive symptoms, with an antidepressant effect maintained at 3 months [25].
Specific concerns also exist in BD antidepressant strategies, such as the manic switch, and so warrant specific safety studies [14]. Depending on the clinical assessment (no systematic report, clinical examination, or rating scales) and clinical characteristics of studied subjects, the rate of manic switches went from 0% to 18.8% [26]. Preliminary reports suggest that morning exposure may induce manic switches, and that mid-day exposures may be associated with a decreased risk of manic switch [27]. A 6-week randomized double-blind placebo-controlled trial provided new evidence to support mid-day BLT as an efficient therapy in BD, but also a safety one since no manic switches were observed [28]. Nevertheless, most of the BD patients (82%) had BD with seasonal pattern, and no comparisons of seasonal and non-seasonal patients were possible. Moreover, whereas an historical review suggested that manic switches occurred independent of treatment modality [26], different dose-titration protocols have not been directly compared and data are lacking in order to provide reliable evidence-based therapeutic guidelines.
In this context, the originality of this study protocol will be to evaluate, in patients with BD and non-seasonal depression, using a rigorous approach, the characteristics of BLT administration that are not codified (duration, escalation, morning and mid-day exposure) depending on the tolerance (hypomanic symptoms) from an intra-subject dose escalation scheme of the efficacy of this therapy, in two groups exposed either during the morning or at mid-day. This phase I/II study will also provide an estimate of the potential size effect of BLT, which is necessary for the secondary planning of a randomized, placebo-controlled efficacy trial.
METHODS
Aims of the study
The objectives of this pilot study in the non-seasonal BD depression are to evaluate the modalities of the therapeutic escalation according to the tolerance of morning and mid-day BLT.
The main objective is to establish the maximum tolerated dose (MTD) of BLT [(hypo)-manic switch] in two parallel groups of BLT: morning or mid-day exposure. Secondary objectives are: 1) identify the optimal duration of acute BLT treatment by analyzing the kinetics of symptoms progression especially at 6 weeks, 8 weeks and 10 weeks for both morning and mid-day BLT, 2) evaluate feasibility and acceptability, 3) characterize the clinical markers associated with BLT response, 4) evaluate the tolerance and its acceptability at 6 months, and 5) test the glasses placebo for future randomized placebo-controlled trial.
Global design of the study
The LuBi study obtained the ethical approval from the french Committees of Protection of Persons (CPP de Ouest 6-CPP 1002-DM2) the 20th of June 2017, and the French National Drug and Health Product Agency (ANSM) authorization from the 12 of December 2017. The study is registered under the ClinicalTrials.gov Identifier: NCT03396744. A Data Safety Monitoring Board (DSMB, independent members from investigators) and a Steering Committee have been identified to follow and secure this research.
After the information and screening visit, patients with bipolar depression, i.e suffering from a major depressive episode with a bipolar disorder subtype I or II, without a seasonal pattern, and already treated by a mood stabilizer will participate. Written informed consent will be obtained from patients recruited from five University-affiliated psychiatric departments in France: Hôpital Fernand Widal–Lariboisière, AP-HP, Paris (Pr Bellivier), Hôpital Louis Mourier, AP-HP, Paris (Pr Dubertret), Hôpital Sainte-Anne, Paris (Pr Gorwood), CHU Montpellier (Pr Courtet), CHU Lille (Pr Vaiva).
Patients will first receive placebo (glasses light of 50 Lux close to ambient light) during 1 week and then will be randomized between two conditions of exposure, either at morning or at mid-day. Then, they will be exposed to active bright light with glasses using a dosage escalation (inter- and intrasubject) scheme for 7.5, 10, 15, 30 up to 45 min for 10 weeks. Finally, patients will be assessed 6 months after this intervention. Only during the 10-week intervention, patients won’t be allow to benefit from other antidepressant strategies.
The study follows the global organization summarized in Figure 1 and will be further detailed.
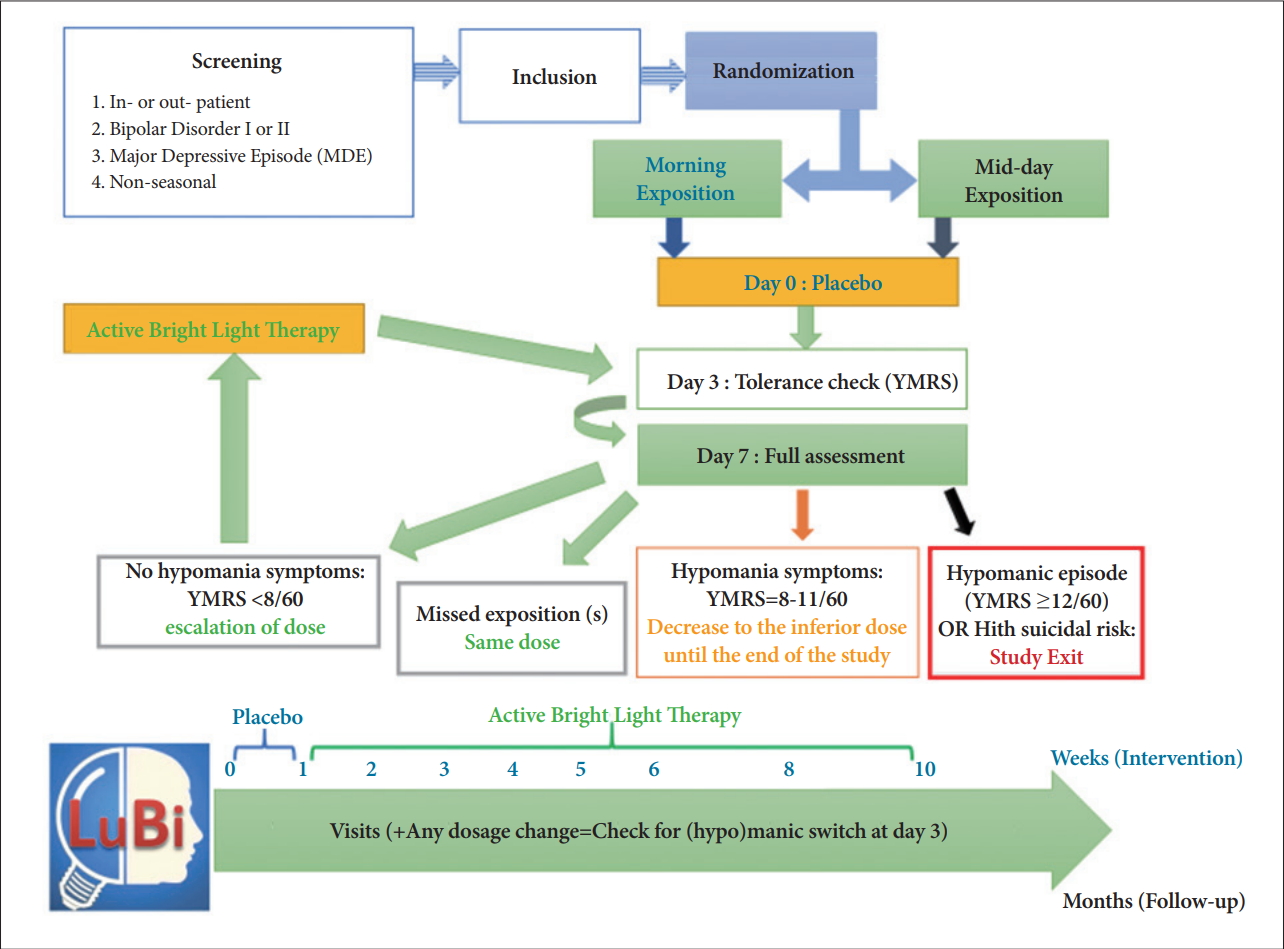
Study design and global organization of the LuBi study. YMRS: Young Mania Rating Scale.
Evaluation criteria
Main criterion
The main criterion will be the dose limiting toxicity (DLT), defined on the hetero-questionnaire Young Mania Rating Scale (YMRS) within 1 week after any dose onset (or change). Any occurrence of a hypomanic switch, based on a total score ≥12/60, or of a sub-syndromic hypomanic symptom (based on a score between 8 and 12), will be accounted for a DLT. The occurrence of DLTs will define both the dose (duration of exposure) of the intra-subject dose escalation (and this regardless of the morning or mid-day group), and the intersubject dose escalation, i.e. the dose of the next cohort of patients (cohorts of 3 patients spaced of at least one week).
Secondary criteria
The secondary criteria were as follows, and all scales are summarized in Table 1:

Summary of all scales used in LuBi and their interpretation
1) Efficacy: decrease in mean depressive score at 6, 8, and 10 weeks as assessed by Montgomery-Åsberg Depression Rating Scale (MADRS) and by the Clinical Global Impressions (CGI).
2) Tolerance (adverse events) with evaluation of the manic switch by the Young Mania Rating Scale (YMRS) hetero-questionnaire at day 3 and then at week 1, 2, 3, 4, 5, 6, 8, and 10, or in case of premature termination of the study.
3) Acceptability of the light glasses assessed by the self-questionnaire qualitative assessment of acceptability designed by the investigators at 1, 2, 3, 4, 5, 6, 8, and 10 weeks.
4) Effects on depression along the follow-up and at 6 months: decreased in mean depression score from 0 to 1, 2, 3, 4, 5, 6, 8, and 10 weeks, and 6 months as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS).
Tertiary criteria
1) Other evaluations with the following rating scales used at 0, 1, 2, 3, 4, 5, 6, 8, 10 weeks and 6 months:
- Clinical Global Impressions (CGI)
- Columbia Assessment Scale on Suicidal Risk Severity (C-SSRS)
- Quick Inventory of Depressive Symptomatology (QIDS SR-16)
- Self-questionnaire of manic symptoms (Altman questionnaire)
- Pittsburg Sleep Quality Index (PSQI)
- Chronotype: Composite Scale of Morningness (CSM)
- Circadian Type Inventory (CTI)
- Daytime sleepiness: Epworth Sleepiness Scale (ESS)
- Side effects: TAKE-M scale
2) Other rating scales used only at baseline:
- Seasonality: seasonal pattern assessment questionnaire (SPAQ)
- Risk of obstructive sleep apnea / hypopnea (OSA): Berlin scale
Eligibility criteria
Inclusion criteria
Inclusion criteria are:
- Patients must be aged from 18 to 55 year-old.
- Patients must read and understand French language, and must provide written informed consent.
- Patients must be inpatients or outpatients followed in psychiatry for a major depressive episode.
- Patients must have a diagnosis of BD type I or II according to DSM-IV criteria and determined by a SCID.
- Patients must have a major depressive episode, at least of moderate intensity, according to the DSM-IV, determined by a SCID, and with a MADRS total score ≥20.
- Patients must have a mood stabilizer since at least 4 weeks at standard dosage: lithium (plasmatic lithium >0.5 mEq/L for instant release (Téralithe 250) and >0.7 mEq/L for slow release (Téralithe LP 400), or sodium valproate (plasmatic dosage >40 mg/L), or second-generation antipsychotics such as quetiapine (dosage ≥150–800 mg), aripiprazole (dosage ≥15–30 mg), olanzapine (dosage ≥10–20 mg).
- Female patients must be using a medically accepted means of contraception.
- Patients must be affiliated to the French social security system.
Exclusion criteria
Exclusion criteria are:
- Patients under guardianship or deprivation of liberty by administrative or judicial decision.
- Seasonal pattern of major depressive episode according to DSM-IV criteria.
- Psychotic, mixed, or catatonic characteristics according to DSM-IV criteria.
- Serious suicidal risk assessed by the Columbia Scale of Suicide Risk Severity (C-SSRS).
- Not stabilized comorbidities (addictive disorders according to the DSM-IV criteria or other decompensated general medical disorder).
- Ophthalmic pathology (cataract, macular degeneration, glaucoma, retinitis pigmentosa) and diseases affecting the retina (retinopathy, diabetes, herpes, etc.).
- Photosensitive treatment, including the following treatments:
• Cyclins (Vibramycine®, Doxycycline®)
• Amiodarone (Cordarone®, Amiodarone®)
• Phenothiazines (Largactil®, Modecate®, Nozinan®)
• Methotrexate (Methotrexate®)
• Sulfamides (antibiotics, diuretics or hypoglycemic agents)
• Chloroquine (Nivaquine®)
• Some anti-inflammatories (Apranax®, Indocid®)
• Psoralens used in puvatherapy
• Isotretinoin (Ruoaccutane® and generics)
• Verteporfin (Visudyne®)
- Lactating or pregnant women (pregnancy urine positive test)
- Subjects who have already used light therapy in the last 6 months.
- Therapeutic resistance of the current major depressive episode (≥2 traditional antidepressants such as SSRI, NaSRI, MAOI or tricyclic, at effective therapeutic dosage for more than 6 weeks)
- Use of another antidepressant strategy than the mood stabilizer, including antidepressants of all classes (which will have to be stopped before the initiation of light therapy) and psychotherapy with onset <1 month.
Light device
Identification of the device
The BLT will be administered using specific glasses (Luminette®). It is a medical device that is produced by the company Lucimed called Luminette® (EAN: 0702382929671), which weighs 0.6 kgs and has the following dimensions: 22×11×11 cm. In addition to the innovative nature of this device, the glasses allow control of the light exposure and good ergonomics (the patient can continue its activities). This device is freely available on the market. The technical details of the Luminette® are available on demand. The device provides daily exposure to a fluorescent light of 10,000 Lux perceived, possessing a UV filter and CE standards. Luminettes® of the study will be blocked on intensity 2/3, a blue enriched-light of 1000 Lux, equivalent to 10,000 Lux on traditional bright light therapy [29]. It has been observed that low-intensity blueenriched light (750 Lux) has a therapeutic effect comparable to standard bright light (10,000 Lux) in treating seasonal affective disorder [30]. A control exposure (Placebo) will also be set up, using the same Luminette® device and CE standards, but offering a fluorescence of 50 Lux, close to ambient light, and therefore considered ineffective. The objective of the placebo is to have a zero-dose measurement of light therapy; it will also allow the patient to get used to the device.
Utilization of the device
The dosage, as indicated above in the study design, will depend on the therapeutic escalation. It is based on the definition of several intra-subject dose escalation schemes, with different initiation dose and a total of 5 possible dose levels (7.5, 10, 15, 30, and 45 min). This intra-subject escalation scheme can be done at most once a week. The modes of administration are the same for the subjects exposed in the morning and those exposed at mid-day, with the wearing of BLT glasses (Luminette®) for a determined duration according to the therapeutic escalation, and a recording of the time of exposure. The patient can continue his usual activities. This administration will be done at a fixed time and common to all centers: 1) BLT in the morning: 8 am±30 min; 2) BLT at midday: 12 hours (noon) ±30 min.
Training and experience required to use the device
All patients will receive by an investigator standardized verbal and written instructions on the setting up of the Luminette®. Adherence will be monitored using daily logs of device treatment times completed by patients and reviewed at each visit. This monitoring of adherence has already been used effectively in recent studies, including a very recent randomized controlled trial of light therapy published in the JAMA Psychiatry [21].
Investigators’ training in the use of the Luminette® device will be done by the principal investigator (Dr. PA Geoffroy) and requires no prerequisite. Training will be organized in each center with the center’s investigators. The use is very simple with a push button to turn on/off the device and an electric charger to recharge after use. The correct placement of the device will be shown using diagrams (Figure 2) and then simulated during training between the investigators. This training in the use of the medical device lasts 15 minutes per trained staff.
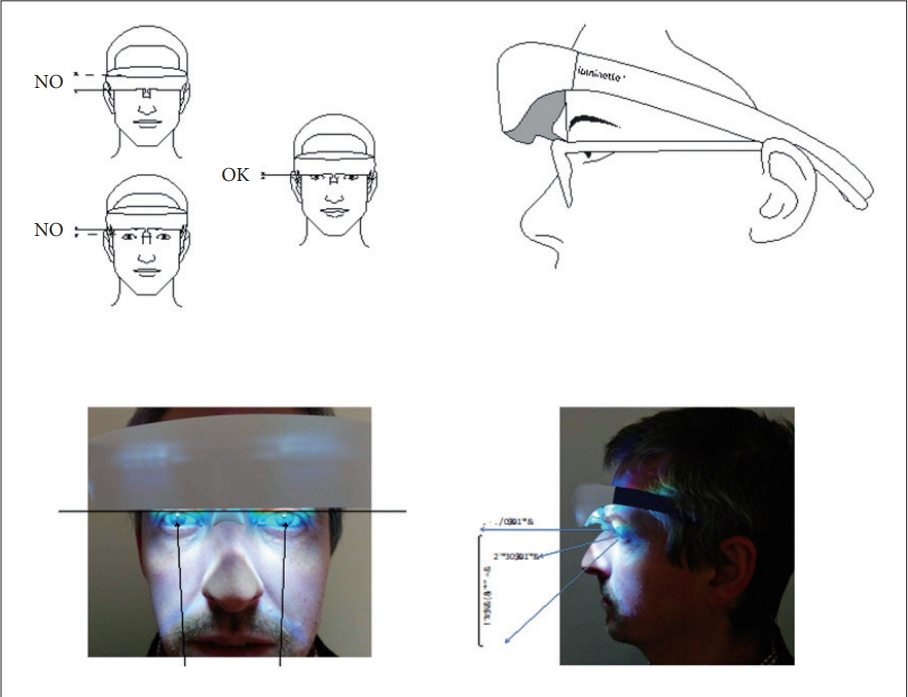
Correct placement of the Luminette® device.
Experimental plan
This is a dose-finding study–the dose being defined as the duration of exposure to light- based on an inter- and intrasubject dose escalation scheme to determine the maximum tolerated dose (MTD) of BLT in Bipolar depression. This dose research will be carried out in two randomly allocated groups in 1:1 : 1) BLT in the morning, 2) BLT at mid-day.
The dose is defined in this study by the duration of the exposure to the intermediate intensity of the BLT glasses. This scheme allows to take into account the inter- and intra-individual variability of tolerance and response, to model cumulative toxicity and to maximize the probability for each patient to receive an effective dose. It is based on inter and intrasubject dose escalation algorithms, with different initiation doses among a total of 5 dose levels (7.5, 10, 15, 30, and 45 min). In the absence of respect by the patient of the duration of exposure to BLT (defined as one day without exposure during a week) during the placebo phase, this will lead to a study termination; during the first week of the active treatment, this will lead to add a new patient to the ongoing cohort, and at any other phases of the study, this will lead for the subject to have the same dose one more week.
Inter-subject or inter-cohorts’ escalation
The inter-subject escalation scheme aims to estimate the maximum tolerated dose (MTD) of the BLT, as measured on hypomanic switch, and will be based on the standard ‘3+3’ design. Patients will be included by cohorts of 3 patients with inclusions delayed of at least 3 days for safety reasons. The first 3 subjects included (Cohort A) will start with the first dose level (7.5 minutes). In case of the absence of any dose limiting toxicity (DLT) of all the 3 patients from the first cohort, the initiation dose of the next cohort will be increased to the next level (i.e. 10 min, Cohort B). To allow the observation of DLT, the dose escalation of the new cohort will be allowed only 1 week after the inclusion of the last patient from the previous cohort. The inter-subject escalation is based on the observation of no DLT on the 3 subjects of a cohort, or at most on 1/6 as if one meet a DLT, a second identical cohort starting at the same initiation dose, and the escalation will be possible only if none of this new supplemental cohort meet a DLT (Figure 3). The MTD is defined if two or three subjects meet a DLT from the same cohort or two cohorts at the same dose initiation (in the case when only one subject meet a DLT in the first cohort, and a second cohort at the same dose initiation must have been started to confirm or not this dose initiation safety). So on for the following cohorts (Figure 3).
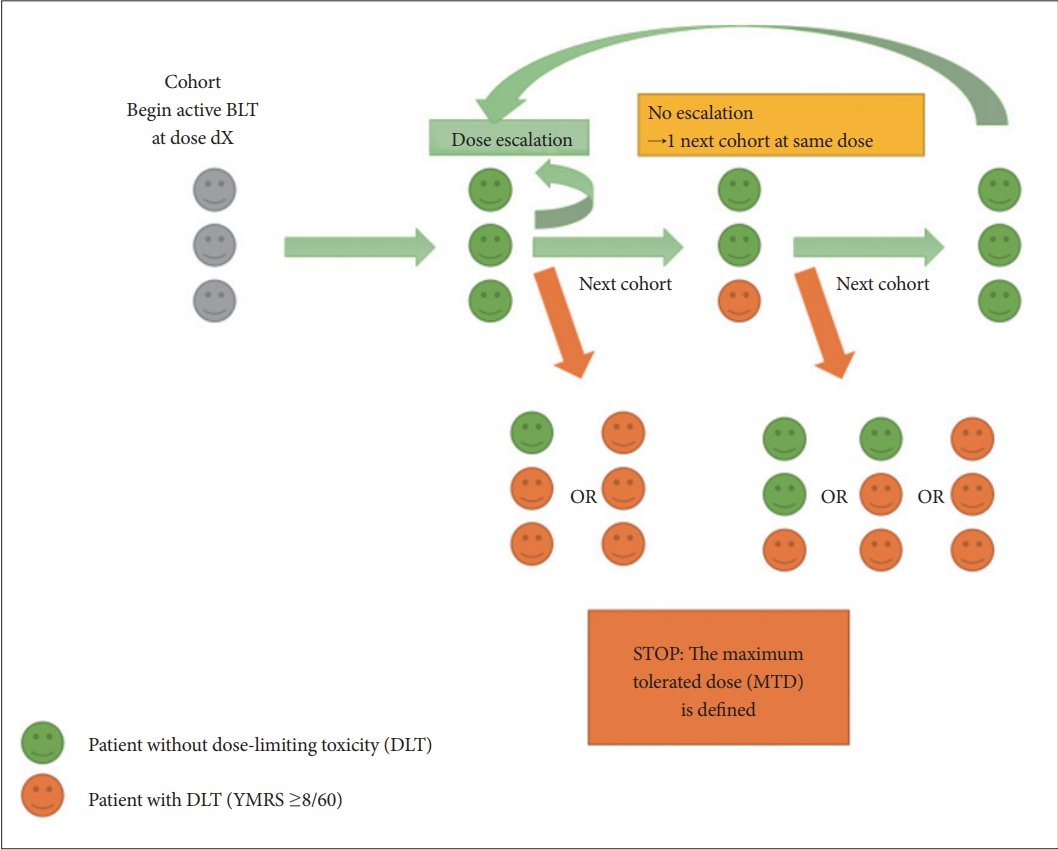
Diagram of the inter-subject or inter-cohorts’ escalation. DLT: dose-limiting toxicity, MTD: maximum tolerated dose, BLT: Birght Light Therapy, YMRS: Young Mania Rating Scale.
Intra-subject’s escalation
The intra-subject escalation scheme aims to account for individual variability in the definition of the MTD [31], allowing each subject to receive the maximum dose that he(she) tolerates. The intra-subject dose escalation will occur only once a week for each patient.
This dose escalation will take into account any DLTs (ie, hypomanic switches) observed during the escalation on the previous cohorts, through the observation of so-called “Target Ceiling Dose” (TCD) [32] (Figure 4).
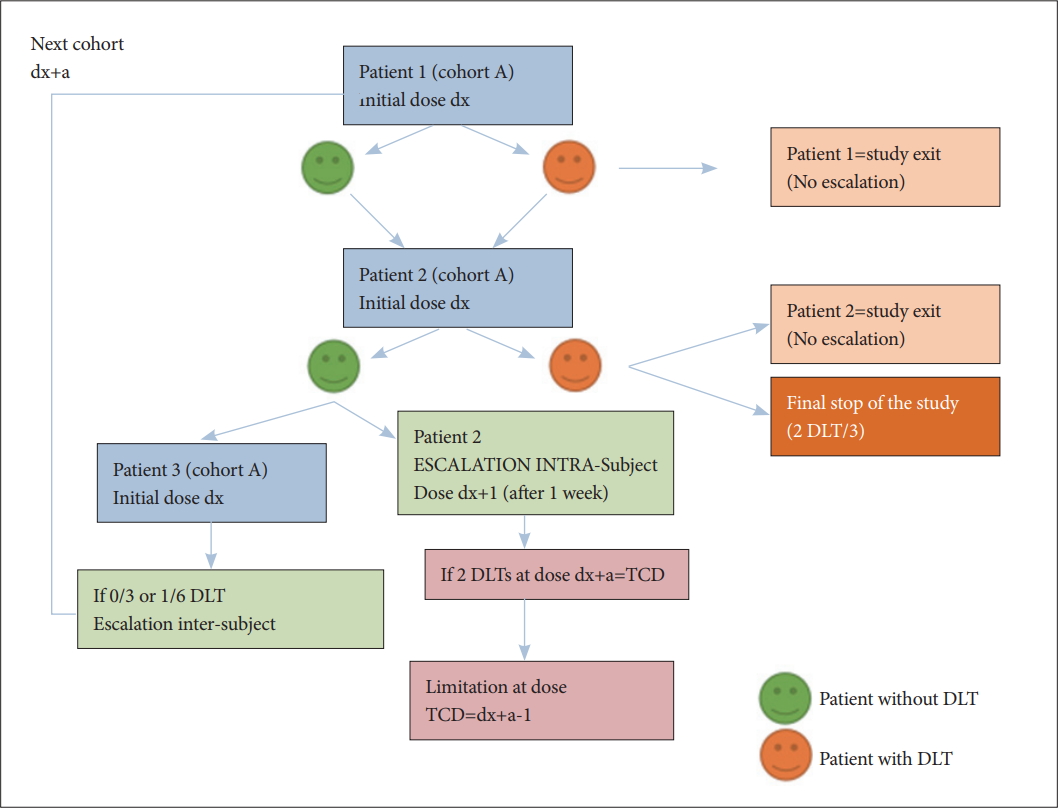
Diagram of the the intra- and inter-subject escalation decision rule for a cohort. The diagram schematizes the intra-cohort and inter-cohort escalation rules for a cohort of 3 patients (number 1, 2, and 3) treated at an initial dose dx. The inter-subject escalation is based on the observation of no DLT on the 3 subjects of this cohort (or at most on 1/6); the intra-subject escalation is performed in the absence of DLT at the initial dose, and it will stop if the subject is experimenting with a DLT at the dose dx+a. Intra-subject escalation or the initial dose for future cohorts will then be limited to the dose immediately below the target ceiling dose (TCD) as soon as it has been reached for at least two patients. DLT: dose-limiting toxicity, TCD: target ceiling dose.
Rules for this escalation can be summarized as follows:
• All patients will start with a first week with a placebo for 10 minutes. This week with placebo aims to have a zerodose measurement of light therapy; it will also allow the patient to get used to the device.
• In the absence of DLT (YMRS score <8), active treatment at the initial dose will be started.
• If the subject has DLT (YMRS score ≥8) within one week after treatment onset, the patient will leave the study with planned management of the mood episode; this doselimiting toxicity (DLT) will be recorded in the rest of the trial for the following cohorts (Figures 3 and 4).
• If this is not the case, there will be an intra-subject escalation of the dose administered on the next week to the upper level (immediately above).
- If YMRS score <8 has been observed in the patient, escalation will be proposed to the higher dose (if available) after 1 week at the same dose.
- If YMRS score between 8 and 12, defining sub-syndromic hypomanic symptoms, the intensity of light therapy will be reduced to the previous level (de-escalation to the lower dose, if it exists, and if it does not exist, this will result in a study termination).
- If YMRS score between 12 and 20, the light therapy will be interrupted and the study will end for the subject with planned management of the thymic episode (see below).
- If YMRS score ≥21, it defines a manic episode (Table 1), this will lead to the same result as a previously described hypomanic episode, namely a study termination with usual psychiatric hospital care.
If a patient presents during the escalation (ie after the initial dose, “dx”) a toxicity or a change in mood at dose level (dx+a, “a” being the escalation dose level), the escalation will be abandoned for this patient and the dose (dx+a-1, “-1” being the decreased of the dose to the lower level) will define the DLT for this patient. If 2 DLTs occurred at a dose dx+a, the “Target Ceiling Dose” (TCD) will be defined -which is the highest theoretical dose that can be administered to a patient 30- and lead to a maximum dose for all participants at a dose below the TCD (dx+a-1), such as the dose level immediately lower (if it exists) than the one that led to a YMRS score ≥8.
Intra-subject escalation will therefore be interrupted at the dose at which the patient exhibits DLT, or when a patient has reached the dose that has been defined as a TCD for two patients in the trial: as soon as 2 patients have reached a DLT, the TCD observed (dx+a) will be applied to patients from the following cohorts.
So this concept of TCD will be used both in the intra-subject escalation and inter-subject escalation (i.e. between two cohorts of 3 patients included at the same dose), which can not start with an initial dose at this TCD (Figure 4). Note that the TCD supported on at least 2 subjects during intrasubject escalation will be communicated to the investigators upon inclusion; eventual updates during trial will also be communicated on the site in real time, with an alert message to the clinicians involved by email.
Organisation of assessements during intra and inter-subject escalation
Each participant will be evaluated at each dose level after 3 days, at 1, 2, 3, 4, 5, 6, 8, and 10 weeks, then at 6 months, and in case of premature discontinuation. Patients will benefit from these assessments either in hospital if they are hospitalized, or in a day care centre for individual with bipolar disorders if their condition does not justify full-time hospitalization. In addition, if the suicidal risk (systematically assessed at 1, 2, 3, 4, 5, 6, 8, and 10 weeks) worsens and becomes serious (evaluated as such by the C-SSRS) (Table 1), it will lead to study termination and routine psychiatric care of the prevention of suicidal behaviors.
The control of the 3rd day makes it possible to ensure early, and before the complete evaluation at 1 week, of the good tolerance of each dose escalation, via the verification of the absence of hypomanic symptoms and suicidal risk as previously described. This control on the 3rd day can therefore lead to a dose de-escalation or a study termination.
The distribution of patients between the different doses of BLT does not depend on randomization, but on the observation of DLT on patients included at previous levels. Randomization is only used to test the effect of BLT conditions, ie morning vs. mid-day. The choice of doses is based on the observed responses of patients previously included in a sequential pattern called “adaptive”. This theoretical distribution can be summarized in the flow chart in Figure 5.
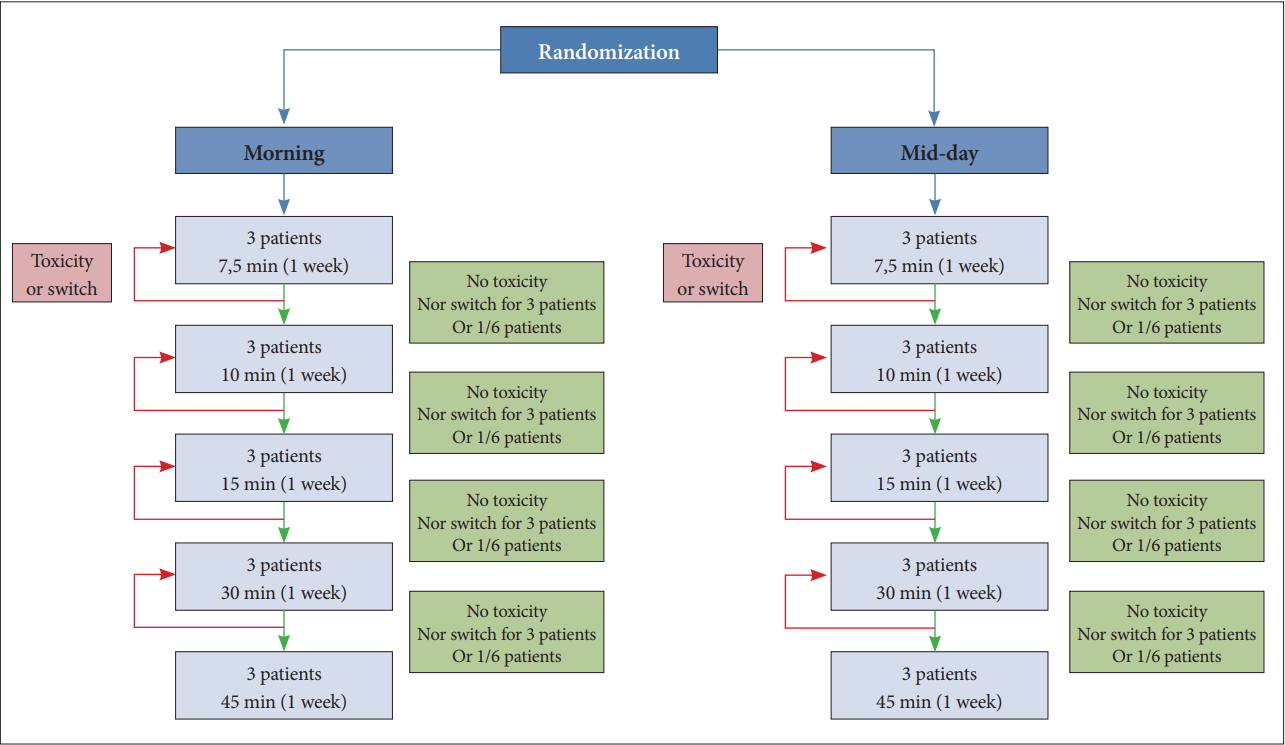
Theoretical distribution of doses between the randomized groups, morning and mid-day. Randomization is only used to test the effect of BLT conditions, ie morning vs. midday. The choice of doses is based on the observed responses of patients previously included in a sequential pattern called “adaptive”. BLT: bright light therapy.
Randomization
Patients will be randomized centrally for 1 of 2 treatment conditions:
1) BLT in the morning
2) BLT at mid-day
For each patient included, the investigator in charge of the patient will be given an anonymous identification number. The randomization into two 1:1 parallel groups will be done after verification of the eligibility criteria (respect of the inclusion and non inclusion criteria). It will use pre-established randomization lists, balanced by permutation blocks, and stratified on the center. It will be carried out via the eCRF (electronic Case Report Form) CleanWeb tool randomization module, centrally, by logging into the eCRF website dedicated to the test. Investigators will need to connect centrally to this site when including patients in the study to find out how to administer BLT (morning or mid-day).
Once randomized into either group, the initial exposure dose of the patient will be transmitted to the physician in charge of the patient.
Conduct of the research
Screening visit
This research protocol will be proposed to any adult patient with a BD type I or II hospitalized or followed in psychiatry for a non-seasonal depressive episode. One of the investigators will provide oral and full written information.
Inclusion visit
The patient inclusion will be done after at least one day after information, and only following the collection of free and informed consent. The inclusion and non-inclusion criteria will be checked and the Structured Clinical Interview for DSM (SCID) will be used, which is a standardized and validated semi-structured tool [33]. Then the centralized randomization of the patient will be done according to the modalities detailed previously.
Participation in the study will be done after the inclusion visit. For the validity of the inclusion criteria, a maximum period of 15 days must be respected between the inclusion visit and the inclusion in the trial.
The criteria for premature termination of inter-cohort escalation are defined by the occurrence of more than one DLT on 3 subjects of the same cohort, or more than 2 on 6 patients (Figure 3). In this case, the MTD is defined by the dose at which these events occurred. This is a definitive stop of escalating doses.
The only temporary stops are related to the need for a delay between inclusions to ensure the absence of toxicity (and avoid the simultaneous inclusion of 3 subjects at the same unexplored dose). Inclusions will therefore be delayed within the same cohort by 3 days. Finally, since the escalation is based on the observation of the response (in terms of oneweek YMRS score) of the 3 subjects included in each cohort, a minimum of one week after the last included cohort will be required before inclusion of a new cohort.
Next visits and follow-up duration
During the therapeutic escalation phase: each participant will be assessed at each dose level after 3 days to ensure the absence of a (hypo) manic switch, and will have a complete assessment at 1, 2, 3, 4, 5, 6, 8, and 10 weeks.
After the 10th week, each participant will stop the daily treatment with active BLT and will be re-assessed, fully, at 6 months.
Therefore, the participation in the study for each participant is 6 months (plus 1 week of placebo), except in case of premature termination.
A patient card with the telephone number of the psychiatric emergencies of each center, will be given to each patient. All assessments during visits are summarized in Table 2.

Summary of evaluations for each visit
Statistics
Statistical power calculation
This is a phase I/II dose finding study with two parallel groups (morning, mid-day) randomly defined (Figure 5). The sample size in these early phase trials is about twenty subjects, based on the 5 possible dose levels for each subject, and 3 subjects per cohort (inclusion per group of 3 subjects), taking into account the possibility of including 3 additional subjects at each dose (in case of DLT), to include at least 15±6 patients per group (morning or mid-day), for a total of 45 patients.
Statistical methods
The statistical analysis will be carried out according to the intention-to-treat principle, ie each included subject will participate in the analysis in the group assigned by randomization, regardless of the “treatment” effectively received, apart from the withdrawal of consents.
The final analysis will be done after inclusion of the total number of subjects, except in case of premature termination of the trial (if the MTD has been reached) where only available data will be used.
A mixed model will allow to model the subject effect (random), the initial dose effect, and cumulative doses. We will estimate the probabilities of success according to these parameters in order to propose an optimal scheme of BLT administration. This dose-toxicity model will therefore incorporate parameters for both intra- and inter-subject variation and cumulative doses [34].
All analyses will be performed on the SAS software (SAS Inc., Cary, NC, USA).
DISCUSSION
Taken as a whole, this study will aim to identify the best modalities of the therapeutic escalation according to the tolerance of morning and mid-day BLT in patients with nonseasonal bipolar depression. In each parallel group (morning vs. mid-day), an inter-subject (with cohorts of 3) and intrasubject (with a potential of 5 dose levels) dose escalation schemes will be used to determine the maximum tolerated dose (MTD) of BLT in BD. The dose is defined here by the duration of the exposure to the intensity of the glasses (equivalent to 10,000 Lux on traditional light therapy). This scheme takes into account the inter and intra-individual variability of tolerance and response, to model cumulative toxicity and to maximize the probability for each patient to receive an effective dose. It is based on the definition of an intra-subject dose escalation scheme, with different initiation doses and a total of 5 dose levels (7.5, 10, 15, 30, and 45 min), and one dose target ceiling dose (TCD) defined according to previous observations during escalation.
This study will propose an antidepressant strategy other than electroconvulsive therapy or antidepressants, in patients who do not respond to well-managed mood stabilization therapy. Light therapy also improves sleep and abnormal circadian rhythms such as phase shift [35-39]. These abnormalities in circadian rhythms and sleep are present in all phases of BD, particularly during depressive phases [40-46].
Concerning the predictive risks, light therapy is rather very well tolerated [47]. More specifically, the possibility exist of hypomanic switch in patients with BD with possible symptoms such as tachypsychia, logorrhea, increased energy and activity, irritability or aggression [27,48]. The other rare side effects of light therapy are: headache, eye strain, nausea and agitation [38,49,50]. In addition, despite traditionnal precautions in BLT studies including what we planned for this LuBi study, there does not seem to be any retinal toxicity associated with light therapy [51]. Regarding the issue of suicidal behavior, which may be a problem with conventional antidepressants, a previous report of 3 cases treated with light therapy alone (2 subjects with BD suffering from seasonal depression and 1 unipolar subject with seasonnal affective disorder) observed in these subjects the emergence of suicidal ideation during the first week, complicated of a suicide attempt for 2 of these 3 subjects [52]. Nevertheless, a more recent study focused specifically on suicidality effect of light therapy in patients with seasonal affective disorder (both unipolar and bipolar) and rather observed a protective effect: almost half of the 191 subjects had a decrease in suicidal ideation and only 6 subjects showed worsening of these [53]. This anti-suicidal protective effect has also been observed in two studies associating light therapy with other therapeutic strategies in unipolar disorder [54] and bipolar disorder [55]. Thus, light therapy may precipitate or aggravate suicidal ideations sporadically, but seems to have a protective effect suicidal in the vast majority of patients.
CONCLUSION
Results from this study would help to better determine how to prescribe light therapy in patients with BD to ensure a good initiation and tolerance, in order to help developing a nondrug therapeutic strategy in a field where significant progress is expected since standard therapies are not very effective outside electroconvulsive therapy. Moreover, this study innovates in the field of light therapy with a device in the form of glasses that may be more ergonomic and acceptable.
Acknowledgements
The study was funded by a grant from Assistance Publique–Hôpitaux de Paris (AP-HP), and the sponsor was Assistance Publique–Hôpitaux de Paris (Délégation à la Recherche Clinique et de l’Innovation).