Linkage and Association Analyses of Schizophrenia with Genetic Variations on Chromosome 22q11 in Koreans
Article information
Abstract
Objective
Chromosome 22q11 has been implicated as a susceptibility locus of schizophrenia. It also contains various candidate genes for which evidence of association with schizophrenia has been reported. To determine whether genetic variations in chromosome 22q11 are associated with schizophrenia in Koreans, we performed a linkage analysis and case-control association study.
Methods
Three microsatellite markers within a region of 4.35 Mb on 22q11 were genotyped for 47 multiplex schizophrenia families, and a non-parametric linkage analysis was applied. The association analysis was done with 227 unrelated patients and 292 normal controls. For 39 single nucleotide polymorphisms (SNPs) spanning a 1.4 Mb region (33 kb interval) containing four candidate schizophrenia genes (DGCR, COMT, PRODH and ZDHHC8), allele frequencies were estimated in pooled DNA samples.
Results
No significant linkage was found at any of the three microsatellite markers in single and multi-point analyses. Five SNPs showed suggestive evidence of association (p<0.05) and two more SNPs showed a trend for association (p<0.1) in pooled DNA association analysis. Individual genotyping was performed for those seven SNPs and four more intragenic SNPs. In this second analysis, all of the 11 SNPs individually genotyped did not show significant association.
Conclusion
The present study suggests that genetic variations on chromosome 22q11 may not play a major role in Korean schizophrenia patients. Inadequate sample size, densities of genetic markers and differences between location of genetic markers of linkage and association can contribute to an explanation of the negative results of this study.
INTRODUCTION
Schizophrenia is a chronic debilitating mental disorder affecting up to 1% of the general population. Evidence from genetic epidemiological studies has suggested that genetic factors play a major role in determining the susceptibility to schizophrenia.12 Modest evidence of linkage with schizophrenia has been reported for several chromosomal loci from genome-wide linkage scans. Chromosome 22q is one candidate genomic region. However, the positive linkage signals yielded from this region are weak and encompass a broad region of chromosome 22q11–13.34567 In addition, linkage was not found in studies that specifically tested this region.8910
Another notable finding related to this region and schizophrenia is that 25–31% of patients with velo-cardio-facial syndrome (VCFS) caused by the microdeletions of 22q11 have early-onset psychotic symptoms.1112 This locus, especially the 1.4–1.5 Mb region of chromosome 22q11, has also been implicated in association studies. The catechol-O-methyltransferase (COMT) gene is a long-known functional candidate gene of schizophrenia that still shows very controversial results from association studies with various populations.13 More recent studies have identified genes for proline dehydrogenase (PRODH)1415 and zinc finger domain DHHC domain containing 8 (ZDHHC8)1617 as candidate schizophrenia susceptibility genes from this locus. Especially, the ZDHHC8 rs175174 polymorphism may function in cortical volume in schizophrenia.18
Given that epidemiologic data on schizophrenia favor the contribution of multiple genes, each exerting a small-to-moderate effect on overall disease risk, controversial results from independent association or linkage studies are only to be expected. Concerning chromosome 22q and schizophrenia, inconsistent results were generated by different populations, even in a single study, which tested combined population samples.91920 To resolve this issue, cumulative data generated by independent investigations of homogenous populations using their own background genetic data and homogenous clinical evaluation system are required.
The purpose of this study was to determine whether genetic variations in chromosome 22q11 are associated with schizophrenia in Koreans. We tested the linkage with schizophrenia using three microsatellite markers in multiplex schizophrenia families. A two-stage case-control association study was also conducted involving an initial screening for 39 SNPs with a 33 kb interval using pooled DNA samples and confirmation analysis using individual genotyping.
METHODS
Subjects
Recruitment of multiplex families containing two or more affected individuals among second degree relatives was performed through proband screening at Samsung Medical Center, Yong-In Mental Hospital, National Chuncheon Hospital and other mental hospitals and psychiatric clinics in Korea. The clinical assessment procedures used were described previously.2122 We limited the diagnosis of the probands to schizophrenia according to the DSM-IV criteria. Affectation status was assigned as broad and narrow liability classes. For the broad class, the affected individuals other than the probands were diagnosed as having schizophrenia, schizoaffective disorder or schizotypal personality disorder according to DSM-IV. In the case of schizoaffective disorder and schizotypal personality disorder, only those cases in which long-term maintenance with antipsychotics was needed were included. The narrow class included those families in which all of the affected individuals met the DSM-IV criteria of schizophrenia. Following the exclusion of one family showing bilineal transmission of psychoses, the clinical data and DNA samples were analyzed from 47 families with 161 members including 105 affected individuals. Among these families, 40 (containing 138 members including 89 affected individuals) also belonged to the narrow class.
For the case-control association study, unrelated schizophrenia patients were recruited from Samsung Seoul Hospital, Yong-In Mental Hospital and National Chuncheon Hospital. Diagnostic Interview for Genetic Studies clinical interviews and best estimate diagnosis were performed by the same raters and psychiatrists who assessed the multiplex families. DSM-IV schizophrenia patients (n=227) were analyzed. Also, 292 normal control persons were recruited and screened for any psychiatric illnesses by means of a brief unstructured interview. The male-to-female ratio of the case and control group was 1.20 and 1.10, respectively (χ2=0.21, df=1, p=0.65). There was no significant difference between the mean age of the case and control groups (33.2±9.6 and 33.5±8.9 years, respectively; t=0.046, p=0.96). The average age at onset of patients with schizophrenia was 25.7±7.0 years (range, 13–49). This study was approved by the institutional review boards of Samsung Medical Center and Yong-In Mental Hospital. Informed consent was obtained from all of the subjects who participated in the interviews and blood sampling.
Genotyping for linkage analysis
Three microsatellite markers spanning a region of 4.35 Mb between 22q11.1 and 22q11.22 were selected (Figure 1) based on their heterozygosities and distances from each other (Table 1). The markers have a 5.86 cM average interval according to the deCODE map. Their heterozygosities were calculated from the genotype data of one-hundred normal Koreans (generated by one of the authors; JWK) and from the unaffected founders of the families of the current study (Table 1). Genotypes were obtained for a broad class of 47 multiplex families with 161 members including 105 affected individuals. DNA was extracted from the white cells of peripheral blood by a Wizard™ Genomic DNA Purification kit (Promega, Madison, WI, USA). In the polymerase chain reaction (PCR), primer sets used were provided by the ABIPRISM linkage mapping set v2.5 (Applied Biosystems, Foster City, CA, USA) for D22S420 and D22S539. For D22S427, the primer set was designed using Primer 3 primer-designing software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3-www.cgi). All of the fluorescently labeled PCR products, by each forward primer, were separated by capillary electrophoresis on an ABI3100 Genetic Analyzer (Applied Biosystems). The results of the automatic scoring of the genotype peaks provided by “Genotyper” program (Applied Biosystems) were manually checked by referring to the distribution patterns of the peaks. Quality control of genotyping utilized DNA sample of CEPH 1347-02 in the assays, with manual checking of the Mendelian transmission for all of the markers and families.
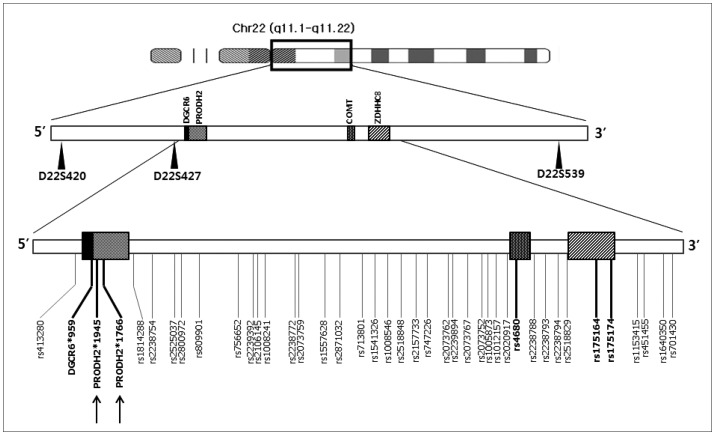
The location of microsatellite markers (spanning 4.35 Mb) and SNPs (spanning 1.40 Mb) used for linkage and association analyses of the present study with known candidate genes of schizophrenia in chromosome 22q11. Intra-genic SNPs on candidate genes are indicated in bold. Arrows represent the SNPs analyzed only by individual genotyping. SNP: single nucleotide polymorphisms.

Results of the single point linkage analysis: non-parametric lod (NPL) scores and pointwise p-values
Genotying for association studies
Initially, an association study using pooled genotyping was done for 39 SNPs (35 intergenic and four intragenic SNPs) with a mean inter-marker distance of 33 kb spanning a 1.4 Mb region of 22q11, which contains four candidate schizophrenia genes (DGCR6, PRODH, COMT, and ZDHHC8) (Figure 1). DNA pools were constructed separately for 227 patients and 292 control subjects as previously outlined.2324 Concentrations of all DNA samples used for pool construction were determined using the PicoGreen dsDNA Quantitation method (Molecular Probes, Eugene, OR, USA) in a Labsystems Ascent Fluoroskan (Life Sciences international, Basingstoke, UK). Three microliters of a DNA suspension (20 ng/uL) from each individual was used to construct DNA pools. Genotyping of DNA pools were performed using SNapShot primer extension method (Applied Biosystems) as previously described23 for each SNP. PCR and extension primers were designed using MassARRAY assay design software (Sequenom, San Diego, CA, USA). Each pooled DNA sample was genotyped four times and average allele frequencies were calculated.
Individual genotyping with all samples included in the pools was performed for SNPs with a trend of association at p<0.1 and four more intra-genic SNPs (Table 2). Genotyping of all the SNPs, except for two on PRODH, were performed with the SNapShot assay (Applied Biosystems). PRODH*1945 T→C and 1766 A→G SNPs were genotyped using the PCR-restriction fragment length polymorphism method. A 499 bp DNA fragment containing the PRODH*1945 T→C polymorphism was amplified using the forward primer 5’-CTCCCTGGTGCGATGGGGTAC-3’ and reverse primer 5’-GGGCCCACACATTCG-AGGAG-3’, followed by digestion with Pvu II (Roche, Basel, Switzerland). The digestion products were scored as cut and uncut type (1945*C allele and 1945*T allele, respectively). A 263 bp DNA fragment containing the PRODH*1766 A→G was amplified using the forward primer 5’-GGGACAG- TGTCCTGGAACTG-3’ and reverse primer 5’-CATCACGGGGCCATAGGGCAC-3’, followed by digestion with Nci I (New England Biolabs, Ipswich, MA, USA). The scored cut and uncut type was 1766*G allele and 1766*A allele, respectively.

Results of association analysis for markers genotyped with individual DNA samples
Statistical analysis: linkage analysis
The model-free linkage analysis was performed using Genehunter 2.1_r4 β.25 Both the single point and multi-point NPLall scores were calculated by an affected-only allele-sharing method. This analysis does not have the context of a genome-wide linkage mapping, and we adopted the pointwise p-value to evaluate the statistical significance. The marker distances obtained from the deCODE map were adopted. The marker allele frequencies were calculated from the Korean population data described above, except for D22S427. For this marker, allele frequencies were calculated from the genotypes of the unaffected founders of the families recruited for the current analysis.
Statistical analysis: association studies
The genotypic distribution of each marker tested for the association studies was not significantly different from that expected based on the Hardy-Weinberg equilibrium in either the case or control group (SAS Genetics v 8.02). In the initial screening with pooled DNA samples, peak heights of the SNaPshot products in a pool of case DNA and in a pool of control DNA samples were measured. The frequency f of allele A was calculated as f(A)=HA/(HA+kHB), where HA and HB are the peak heights of the SNaPshot products representing alleles A and B. A correction factor (k), the ratio of the peak heights in the heterozygous sample (HA/HB), was used to correct for systematic unequal representation of alleles that occurred with most genotyping methods. This was estimated from 8–16 independent heterozygotes samples that were individually genotyped. The frequency f of allele B was calculated as 1-f(A). Estimated allele frequencies were converted to numbers and were tested for approximate statistical significance by χ2 analysis. For individual genotyping methods, genetic associations were also tested using Pearson χ2-test with the SAS statistical software package version 8.2 (SAS, Cary, NC, USA).
Statistical power analysis
The sample size of 80% power is used to avoid false negative associations in genetic association studies. The effective sample size and statistical power were computed assuming 1% prevalence rate of schizophrenia, 1 of D-prime (Linkage disequilibrium), odds ratio of 1.05–1.5 for a risk allele, 1:1 case-control ratio. User defined type I error rate was calculated for the 11 tested SNPs. Genetic Power Calculator software26 was used for the statistical power determination.
RESULTS
Linkage analysis
Mendelian inconsistency was detected in only one family. The family was excluded from the statistical analysis. Forty-six families with 157 members including 103 affected individuals were finally included in the statistical analysis. No significant linkage was found at any of the three markers in single and multi-point analyses (Table 1).
Association study
In the initial analysis with pooled DNA, five SNPs showed positive associations with schizophrenia: rs2073762 (p=0.01), rs2073767 (p=0.0041), rs4680 (p=0.0133), rs1153415 (p=0.0287) and rs701430 (p=0.0068) (Figure 2). Among these SNPs, rs4680 was located in the COMT gene and the others were inter-genic SNPs. Two more inter-genic SNPs showed a trend for association (p<0.1), i.e., rs1008241 (p=0.0656) and rs2073752 (p=0.0883). For those seven SNPs and four additional SNPs on PRODH and ZDHHC8, genotyping with individual DNA samples were performed. In the association analysis, none of the SNPs showed significant association with schizophrenia (Table 2). The statistical power analysis showed the sample size was too small to achieve adequate statistical power. For instance, the statistical power to identify the evidence for association between rs207362 and schizophrenia after the Bonferroni correction under the assumptions of odds ratio for TT genotypes assuming a recessive model (1.44), risk allele frequency (41% LD between the marker allele and the risk allele=1), disease prevalence (1%) was estimated as 11.69%. At least 1070 cases and 1391 controls were required to achieve 80% statistical power under the same assumption.
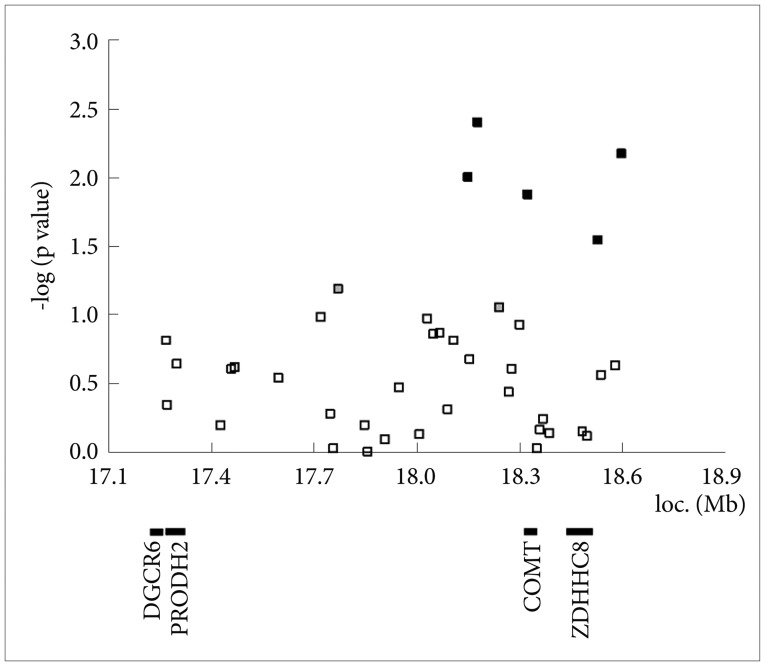
Results of association analysis with pooled DNA samples. Among 39 SNPs, five showed positive association with schizophrenia (black squares) and two showed only a trend of association (gray squares). SNP: single nucleotide polymorphisms.
DISCUSSION
Presently, to assess chromosome 22q11 region as a candidate region for schizophrenia, we describe a linkage strategy and association analysis with a two-stage design. The latter analysis, in which positive marker loci from pooling study are followed by confirmatory individual genotyping, might represent the best trade-off between the cost savings of pooling and the full information that is provided by individual genotyping.
The 22q11 microdeletions have been postulated to be the most prominent genetic risk factor for the development of schizophrenia27 based on their increased prevalence in the disorder. Chromosome 22q11 is therefore believed to be a region that harbors susceptibility genes to schizophrenia, which is also supported by the results of independent linkage studies. Significant linkage score for several populations was reported beginning in 1994.35928 However, other studies reported negative linkage to schizophrenia for this locus.810
Association studies have also been inconclusive. COMT, PRODH, ZDHHC8 and DGCR6 have been suggested as candidate genes in the development or heightened risk of schizophrenia. Thus, in the current two-stage association study we tried to include polymorphisms on those genes. However, our findings are consistent with prior studies that also failed to detect disease association at the COMT rs4680,2330 PRODH* 1945 polymorphism,313233 and ZDHHC8 rs175174.343536
Within all the intragenic SNPs examined in the pooling analysis, only rs4680 on COMT showed a positive trend for association. However, we failed to show same result for in the individual genotyping. Several polymorphisms of COMT have been reported on the significant region; one is rs4680 SNP, a functional Valine to Methionine substitution on codon 158 (Val158Met).133638 In addition, several functional analyses between the polymorphism and schizophrenia have reported significant results. Chen et al.39 showed that the polymorphism affects the activity of COMT with the Met allele having lower activity. More recently, Ohnishi et al.40 suggested that this polymorphism might contribute to morphological abnormalities in schizophrenia.
PRODH has been suggested to increase susceptibility to schizophrenia because of abnormal findings in a PRODH-mutant mouse,4142 350-kb deletion that includes PRODH,43 an allelic association with schizophrenia1544 and identification of PRODH*1766/1945 haplotype that is significantly associated with transmission disequilibrium.44
ZDHHC8 has been also proposed to increase liability to schizophrenia based on both an animal model and human association studies investigating rs175174(A/G) and schizophrenia.17 In addition, no association between schizophrenia and this SNP with overall /divided samples by gender have been reported.3445 In an integrated analysis of multiple datasets, the ZDHHC8 rs175174 polymorphism was negatively associated with schizophrenia.46 These inconsistent findings with regard to the risk allele at rs175174 highlight the need for further studies of the functional effect of this SNP.
The interaction of contiguous genes on 22q11 might influence the etiology of schizophrenia, with 22q11-associated schizophrenia having the characteristics of a contiguous gene syndrome. Synergistic interaction among physically linked genes, which disrupts neuronal homeostatic plasticity, could in principle lead to the high disease risk associated with this locus and/or modulate the expression of the phenotype.41 Whether this hypothesis is true or not, it suggests that the interaction of multiple genes, each with a small-to-moderate effect, can pose an appreciable overall disease risk.
Unfortunately, our study found no evidence of linkage and association between schizophrenia and 22q11-candidate genes from both family-based and case-control samples in Koreans.
Limitations of this study should be kept in mind. First, the chromosomal region investigated was not narrow. It was 4.35 Mb and only three microsatellite markers were used for linkage study. In addition, the SNPs were not close enough to linkage disequilibrium with those microsatellite markers. Second, it seems that the linkage study and association study investigated different chromosomal area, even though they are in 22q11. The regions for the linkage study and associations study did not overlap in terms of genetic analysis. Third, the sample size was too small to achieve adequate statistical power.
In conclusion, the findings suggest that genetic variations on chromosome 22q11 may not play a major role in schizophrenia in Koreans. Inadequate sample size, densities of genetic markers and differences between location of genetic markers of linkage and association can contribute to an explanation of the negative results of this study.
Acknowledgments
This study was supported by grants from the Yongin Psychiatric Research Institute and the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (A040042). We would like to thank Prof. Ji Wan Park and doctoral candidate Eun Pyo Hong in the Department of Medical Genetics at Hallym University College of Medicine for their contribution of statistical power determination.