The Effect of Initial Duloxetine Dosing Strategy on Nausea in Korean Patients with Major Depressive Disorder
Article information
Abstract
Objective
To assess the relative severity of nausea in patients from Korea with major depressive disorder (MDD) who were treated with duloxetine at low (30 mg) or high (60 mg) doses, with or without food, for the first week of an 8 week treatment.
Methods
Adult patients (n=249), with MDD and a 17-item Hamilton Rating Scale for Depression (HAMD17) score of ≥15, received open-label once daily duloxetine. At Week 0, patients were randomized to 4 groups: 30 mg with food (n=63), 60 mg with food (n=59), 30 mg without food (n=64), and 60 mg without food (n=63). At Week 1, all patients switched to duloxetine 60 mg for 7 weeks. The primary outcome measure was item 112 (nausea) of the Association for Methodology and Documentation in Psychiatry adverse event scale. Effectiveness was assessed by change in HAMD17 total score.
Results
Overall, 94.4% (235/249) of patients completed Week 1 and 55.0% (137/249) of patients completed the study. For Week 1, nausea was significantly less severe for patients who received 30 mg compared with 60 mg duloxetine (p=0.003), regardless of food intake. In all groups, nausea severity was highest at Week 1 and declined throughout the study. HAMD17 score was reduced in all groups and the most common adverse event reported was nausea (145/249; 58.2%).
Conclusion
To minimize nausea, Korean patients with MDD who require duloxetine treatment could be given 30 mg once daily, regardless of food, for the first week followed by 60 mg once daily for the course of therapy.
INTRODUCTION
Duloxetine, a serotonin-norepinephrine reuptake inhibitor, is an effective first-line treatment for patients with major depressive disorder.1 Nausea is the most common adverse event reported during acute phase treatment, and is the most common adverse event leading to discontinuation, of patients with major depressive disorder treated with duloxetine.2 The findings from clinical trials suggest that the incidence of nausea is higher in patients from East Asia [37%; 60 mg once daily (QD)]3 compared with Caucasian patients (14.4%, pooled analysis4; 23.4%, pooled analysis5; 29.7%, 60 mg QD6).
Most cases of nausea occur early in duloxetine treatment (within the first 5 days) and are mild or moderate in severity.4,7 The incidence of nausea in Caucasian patients can be reduced by decreasing the dose of duloxetine8,9 or by taking duloxetine with food.10 However, the possible benefits of reducing the initial dose of duloxetine or taking duloxetine with food on the incidence and severity of nausea have not been studied in patients from Asia.
The primary objective of this study was to assess the relative severity of nausea in patients from Korea with major depressive disorder who were treated with duloxetine at low (30 mg QD) or high (60 mg QD) doses, with or without food, for the first week of an 8 week treatment. The secondary objectives of this study included assessment of the effectiveness of duloxetine and additional measures of safety and tolerability.
METHODS
Study design
This phase IV, open-label, randomized clinical trial was conducted at multiple clinical practice sites in Korea between August 2009 and April 2011. The study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and with Good Clinical Practice guidelines in Korea. Institutional ethics approval was obtained before the study commenced. The study was conducted in compliance with the ethical principles of the Declaration of Helsinki, and all patients (or their representatives) gave voluntary, signed informed consent before any study-related procedures or administration of the study drug. The trial was registered at ClinicalTrials.gov: registration #NCT00960986.
Study population
The inclusion criteria for patients in the study included the following: ≥18 years of age; a diagnosis of major depressive disorder (as defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision); a 17-item Hamilton Rating Scale for Depression (HAMD17) score11 of ≥15 at screening and Week 0, and a negative pregnancy test.
The exclusion criteria for patients in the study included the following: a lack of response of the current episode of major depression to 2 or more courses of antidepressant therapy; a current primary Axis 1 disorder (other than major depressive disorder); previous diagnosis of bipolar disorder, schizophrenia, or other psychotic disorders; a serious suicidal risk in the opinion of the investigator and/or score ≥3 on item 3 (suicide) of the HAMD17; an acute or chronic illness with main symptoms of nausea or gastrointestinal discomfort; or taking medication known to have gastric effects that would interfere with nausea ratings.
Study protocol
All patients received open-label QD oral duloxetine (duloxetine hydrochloride; Cymbalta®, Eli Lilly and Company, Indianapolis, IN, USA). The study was divided into 4 study periods (Figure 1): study period I, screening; study period II, 1-week initial dosing period; study period III, 7-week therapy period; and study period IV, optional tapering period. At Week 0, patients were randomized, by a computer-generated random sequence using an interactive voice response system, to 4 treatment groups in a 1:1:1:1 ratio. The treatment groups were (i) 30 mg QD with food, (ii) 60 mg QD with food, (iii) 30 mg QD without food, and (iv) 60 mg QD without food. Patients in the without food groups were instructed not to take duloxetine within an hour of eating. At Week 1, all patients were switched to duloxetine 60 mg QD. During the 7-week therapy period, duloxetine was taken without regard to food and assessment visits occurred at Weeks 2, 4, 6, and 8. At the discretion of the investigator, patients could receive 30 mg QD duloxetine for 7 to 14 days before discontinuation at the end of the study (Week 10). Primary and secondary outcome measures and additional safety evaluations were assessed during the initial dosing period and the therapy period (from Week 0 to Week 8).
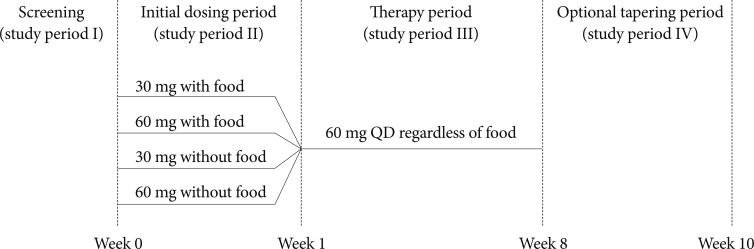
Study protocol of duloxetine treatment. QD: once daily.
Outcome measures
The primary outcome measure was item 112 (nausea) of the Association for Methodology and Documentation in Psychiatry adverse event scale (AMDP-5).12 Mean nausea severity was assessed and time to onset and resolution of nausea were calculated. The secondary outcome effectiveness measures were improvement in HAMD17 total score, subscores and individual items, the Clinical Global Impressions of Severity (CGI-Severity) scale,13 and the Patient Global Impression of Improvement (PGI-Improvement) scale. Response was defined as a ≥50% reduction in HAMD17 total score from Week 0 to Week 8. Remission was defined as a HAMD17 total score of ≤7. Treatment adherence, as defined by the number of capsules taken versus the number of capsules prescribed, was also assessed. Patients were considered to be adherent if, for a particular visit, 80% to 120% of prescribed capsules were taken. Additional safety evaluations included the following: AMDP-5 gastric events score [mean of items 112 and 113 (vomiting)]; AMDP-5 common adverse events score [items 111 (dry mouth)+115 (constipation)+118 (dizziness)+105 (drowsiness)+122 (increased perspiration)+106 (decreased appetite)+mean items 101 to 104 (insomnia)+gastric events score]; individual AMDP-5 items; the frequency and nature of treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs); vital signs; concomitant medications; and the Columbia Suicide Severity Rating Scale (C-SSRS) score (which was only completed when the investigator suspected that an adverse event represented a suicidal thought or behavior).
Statistical analysis
The primary objective included the graphical (non-model based) assessment of the relative severity of nausea. A sample size of 240 patients, allocated in a 1:1:1:1 ratio to the 4 treatment groups, was considered sufficient for graphical assessment and provided adequate power for secondary objectives.
All analyses were conducted on the intent-to-treat population (ITT; all randomized patients, regardless of treatment or protocol deviations). A 2-tailed α level of 0.05 was specified. All appropriate baseline data, safety data, and effectiveness data were summarized using descriptive statistics. Unadjusted mean nausea scores and corresponding 95% confidence intervals were computed for the primary analysis. A supporting factorial analysis using analysis of convariance (ANCOVA) was also performed with fixed effect terms for baseline score, investigator, age, gender, dose group, food group, and dose-by-food group interaction.
The time-to-onset/resolution of nausea was calculated using a life test model producing Kaplan-Meier plots and log rank tests. Treatment differences for the secondary outcomes were assessed by least squares (LS) mean change from baseline using a mixed-effects model with repeated measures (MMRM). Each MMRM included the fixed, categorical effects of treatment group, site, visit, treatment-by-visit interaction, and gender, and the continuous, fixed covariates of baseline score, baseline score-by-visit interaction, and age. The proportion of patients achieving response or remission, or reporting an adverse event were compared using Fisher's exact test. Baseline body mass index (BMI) scores were split into thirds to form the BMI subgroups. All analyses were conducted using SAS® Version 9.1.3 (SAS, Cary, NC, USA).
RESULTS
Participant flow through the study
A total of 311 patients were screened for the study. Of these, 249 patients were randomized (ITT population), 235 (94.4%) completed the initial dosing period, and 137 (55.0%) completed the therapy period (Figure 2). There were 137/249 (55.0%) patients who had at least 1 protocol violation during the study. The reasons for protocol violation included inclusion/exclusion criteria violations (46/249, 18.5%), prohibited concomitant medications (47/249, 18.9%), adherence violations (57/249, 22.9%), and violations that were specific to the study (51/249, 20.5%). The most common reason for discontinuation from the study was adverse events (59/249, 23.7%). The most common adverse events leading to discontinuation were gastrointestinal disorders (37/249, 14.9%), specifically nausea (33/249, 13.3%), and nervous system disorders (14/249, 5.6%), including sedation, headache, dizziness, and tremor.
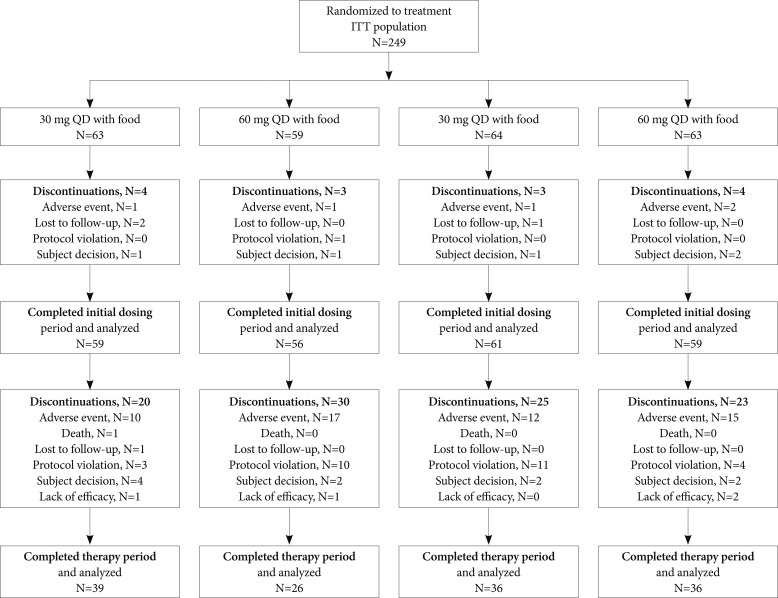
Participant flow diagram. QD: once daily.
Demography and baseline clinical characteristics
Demographic and baseline disease characteristics were similar among the groups (Table 1). Patients were predominantly female (177/249, 71.1%), had a median age of 46.5 years (minimum 20, maximum 79 years), and had their first episode of major depressive disorder about 2 years before enrolling in the study. Patients had a mean HAMD17 total score of 21.6, a median CGI-Severity score of 4.0, and little nausea [AMDP-5 item 112 (nausea) median score of 0, mean score of 0.2].
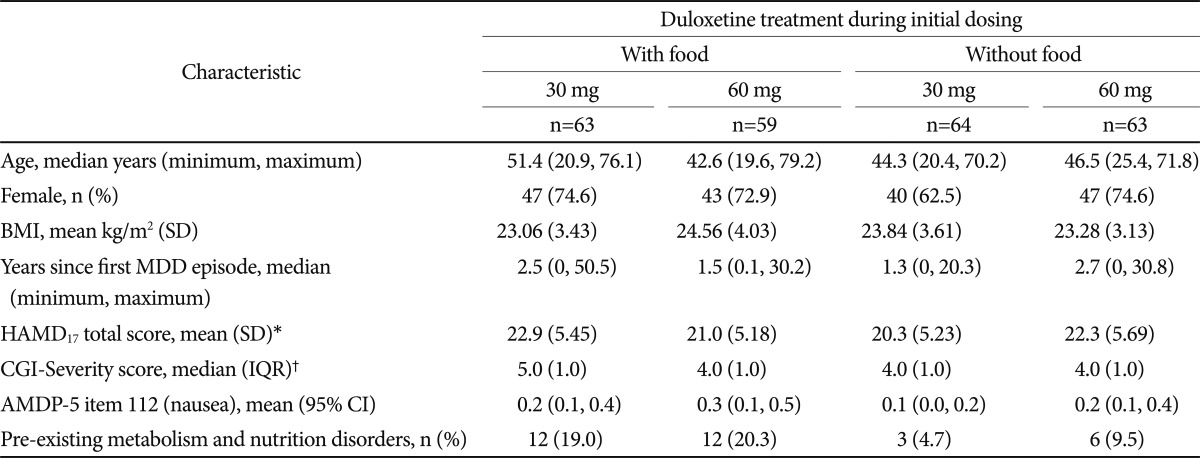
Baseline characteristics
Of note, there were differences between groups (lower incidence in the groups without food, Table 1) for pre-existing metabolism and nutrition disorders, including decreased appetite and hyperlipidemia, but subsequent sensitivity analyses indicated that this variable did not affect the primary outcome.
Nausea incidence and severity
During the initial dosing period, 59.6% (140/235) of patients experienced nausea of any severity. During this period, nausea was significantly less severe for patients who received 30 mg duloxetine (fixed effect of dose, p=0.003), regardless of food intake (fixed effect of food, p=0.555; food-by-dose interaction, p=0.570) (Figure 3). The mean severity of nausea (AMDP-5 item 112 score) was 0.9 and 0.8 for patients in the 30 mg duloxetine groups and 1.4 and 1.2 for patients in the 60 mg duloxetine groups (Figure 3, top left panel). In addition, a higher proportion of patients experienced none or mild nausea in the 30 mg duloxetine groups (about 80%) than in the 60 mg duloxetine groups (about 55%; Figure 3, bottom left panel).

Severity of nausea during initial dosing and therapy periods. For top panels, mean (95% CI) severity by Association for Methodology and Documentation in Psychiatry adverse event scale (AMDP-5) item 112 (nausea) is presented. Fixed effect of dose, p=0.003. Fixed effect of food, p=0.555. For bottom panels, the percentage of patients with each nausea severity is presented.
During the therapy period (Week 1 to Week 8), 39.5% (68/172) of patients experienced nausea of any severity. During this period, the mean severity of nausea (AMDP-5 item 112 score) was ≤0.7 for all groups, regardless of the dose or food intake received during the initial dosing period (Figure 3, top right panel). In addition, the proportion of patients who experienced moderate or severe nausea was low (<20%) in all groups (Figure 3, bottom right panel).
In all groups, the severity of nausea was highest during the initial dosing period (Week 1) and declined throughout the study (Figure 4). By Week 4, the mean severity of nausea was similar to baseline levels and remained low until the end of the therapy period (Week 8). The median time to onset of nausea was longer for 30 mg duloxetine groups than for 60 mg duloxetine groups (30 mg with food, 7 days; 30 mg without food, 6 days; 60 mg with food, 2 days; 60 mg without food, 1 day). For patients who experienced nausea, the median time to resolution of nausea was 8 days (95% CI, 7 to 10 days).
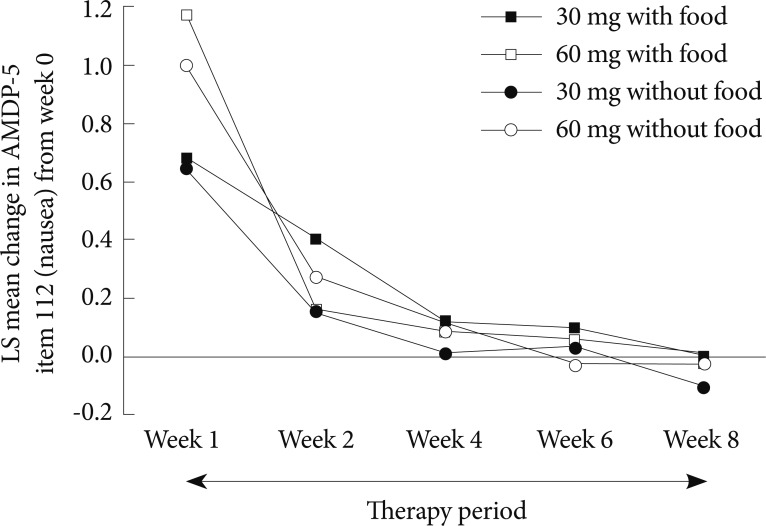
Change in severity of nausea after the patients had switched from the initial dosing period to the therapy period. The least squares (LS) mean change in Association for Methodology and Documentation in Psychiatry adverse event scale (AMDP-5) item 112 (nausea) score is presented. LS: least squares.
Influence of patient age on nausea
The incidence and severity of nausea did not appear to be influenced by the age of the patients. The mean age of patients who completed the study (n=137) was 47.5 years and the mean age of patients who discontinued the study because of nausea (n=33) was 47.7 years. In addition, patient age did not appear to influence the change in nausea that patients experienced from Week 0 to Week 1 (data not shown).
Influence of patient BMI on nausea
The incidence and severity of nausea was influenced by the BMI of the patients. Patients in each BMI subgroup had similar rates of severity of nausea during the initial dosing period (Figure 5). However, the rates of discontinuation because of nausea were higher in the lowest (13/78; 17%) and middle BMI subgroup (13/79, 16%) than in the highest BMI subgroup (5/78, 6%). Mean BMI was similar for males (23.9 kg/m2) and females (23.5 kg/m2).
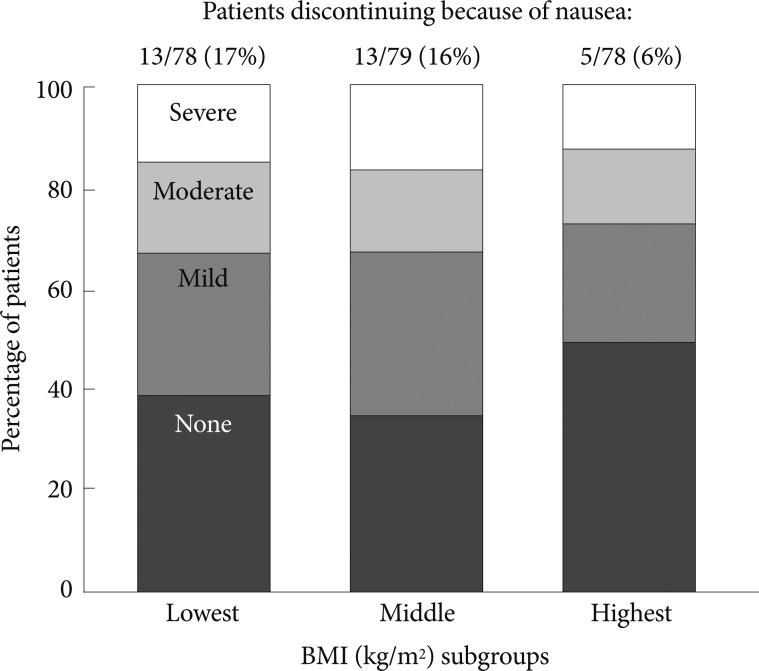
Body mass index (BMI), nausea severity, and discontinuation because of nausea.
AMDP-5 scores
For the AMDP-5 gastric events and common adverse events scores, 30 mg duloxetine was better tolerated than 60 mg duloxetine (Figure 6). The LS mean change in AMDP-5 gastric events or common adverse events score during the initial dosing period (from Week 0 to Week 1) was significantly lower in the 30 mg groups compared with the 60 mg groups (Figure 6). There were no notable clinically important changes in other AMDP-5 individual items (data not shown).

Association for Methodology and Documentation in Psychiatry adverse event scale (AMDP-5) gastric events and common adverse events scores. The least squares (LS) mean (standard error) change in severity by AMDP-5 gastric events score (average of item 112 and 113 (vomiting)] and common adverse events score during the initial dosing period is presented. *p<0.05 compared with 30 mg with/without food. For the gastric events scores: 30 mg with food versus 60 mg with food, p=0.006; 30 mg with food versus 30 mg without food, p=0.702; 30 mg without food versus 60 mg without food, p=0.006; 60 mg with food versus 60 mg without food, p=0.916. For the common adverse events scores: 30 mg with food versus 60 mg with food, p=0.027; 30 mg with food versus 30 mg without food, p=0.725; 30 mg without food versus 60 mg without food, p=0.009; 60 mg with food versus 60 mg without food, p=0.955.
Adherence
Most patients (189/249; 78.4%) were adherent with the treatment throughout the study.
Effectiveness
The HAMD17 total score (and subscores and individual scores, data not shown) significantly improved in all groups (p<0.01 within group) regardless of dose or food intake, with no significant differences between groups (Figure 7).
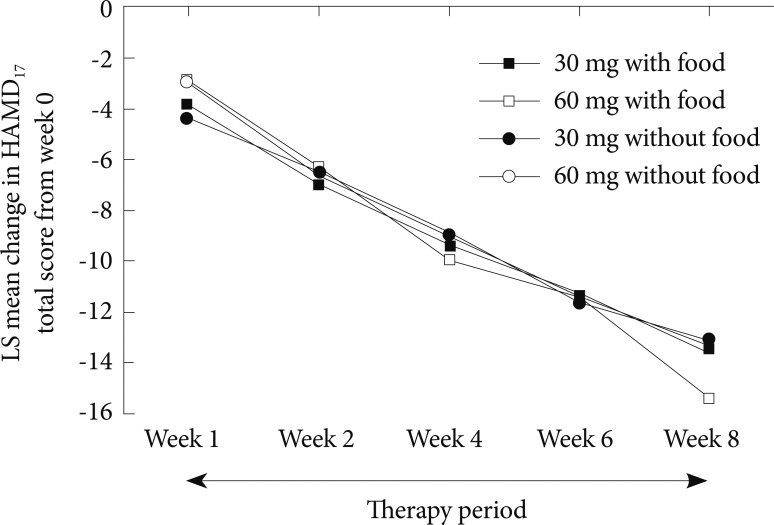
Least squares (LS) mean change in the 17-item Hamilton Rating Scale for Depression (HAMD17) total score after the patients had switched from the initial dosing period to the therapy period. Note there are no statistically significant differences between the groups. LS: least squares.
A total of 115/235 (48.9%) of patients achieved a response and 87/235 (37.0%) achieved remission following duloxetine treatment. The rates of response and remission were similar between groups (data not shown).
The CGI-Severity score significantly improved in all groups (p<0.01 within group) and the PGI-Improvement score significantly improved over time (p<0.001 by visit) regardless of dose or food intake (data not shown).
Adverse events and general safety
Most patients (216/249, 86.7%) reported a TEAE during the study. The most common TEAEs reported were from the system organ classes of gastrointestinal disorders (178/249, 71.5%) and nervous system disorders (94/249, 37.8%). The most common TEAEs reported were nausea (145/249, 58.2%), decreased appetite (46/249, 18.5%), dry mouth (41/249, 16.5%), constipation (39/249, 15.7%), and headache (33/249, 13.3%). Most patients (185/249, 74.3%) reported a TEAE during the study that was considered by the investigator to be related to duloxetine.
There were few differences between groups regarding the nature or frequency of TEAEs. Of note, we observed a difference in the incidence of vomiting between the two 30 mg groups (3.2% with food versus 12.5% without food; p=0.096), however it is uncertain if this was caused by a protective effect of food [the incidence of vomiting in the 60 mg groups was 11.9% (with food) and 14.3% (without food)].
Few patients (4/249; 1.6%) reported a SAE during the study. Although there were no instances of suicidal ideation, suicide attempts or related events from any patients within the study as measured by adverse events and the C-SSRS (data not shown), there was 1 death during the study (in the 30 mg with food group) due to suicide. Other SAEs included diarrhea, pelvic inflammatory disease, muscle injury, road traffic accident, lipoma, and ovarian cyst. All SAEs were considered by the investigator to not be related to duloxetine.
There were no notable changes in vital signs for any patients during the study (data not shown). Concomitant medications were taken by most patients (187/249, 75.1%) during the study, at similar rates for each group. The most common concomitant medications were alprazolam (40/249, 16.1%; for anxiety), lorazepam (70/249, 28.1%; for anxiety), and zolpidem (58/249, 23.3%; for insomnia). Emetogenic medication (with capacity to induce vomiting) was taken by 77/249 (30.9%) patients.
DISCUSSION
This is the first randomized clinical trial located in Korea to investigate the effect of different duloxetine dosing strategies on nausea in patients with major depressive disorder. Gastrointestinal tolerability to duloxetine (based on nausea severity and vomiting incidence) was improved in Korean patients who received a low dose (30 mg QD) compared with a high dose (60 mg QD) during the first week of treatment, regardless of food intake.
In this study, a lower starting dose of duloxetine decreased the incidence and severity of nausea, which is in agreement with findings from other studies. For example, Mallinckrodt et al. found that the incidence of nausea was lower for patients on a dose of 20 mg twice daily compared with patients on a dose of 60 mg QD (16.4% versus 37.8% respectively).9 In addition, Dunner et al. found that the incidence of nausea during the first week of treatment was significantly lower for patients on a dose of 30 mg QD compared with patients on a dose of 60 mg QD (16.4% vs. 32.9%, respectively; p=0.03).8 However, in contrast to Whitmyer et al.10 who found that the severity of nausea was lower when duloxetine was taken with food than without food (p=0.01, regardless of dose), the intake of food with duloxetine during the first week of treatment in this study did not influence the incidence or severity of nausea. As the clinical trial described in Whitmyer et al. was conducted with patients in the United States, the majority of whom were white, it is possible that the difference in the effect of food intake may have been influenced by the differences in diet between the United States and Korea, other differences related to ethnicity, or differences in patient characteristics such as BMI.
The overall incidence of nausea in this study (58.2% by adverse event) was higher than the incidence reported in blinded studies with patients from East Asia (37.1%)3 or Caucasian patients (29.7%6; 14.4% pooled population4). This difference in the incidence of nausea may be because of a decrease in tolerability to duloxetine in the patients from Korea enrolled in the current study. However, it is possible that a higher rate of nausea was reported because the study was focused on nausea. This may also explain why discontinuation because of nausea in the current study was higher (13.3%) than expected.3
The age of the patients in this study did not influence the incidence of nausea or the proportion of patients who discontinued because of nausea. This finding is supported by a pooled analysis of patients treated with duloxetine that found no difference in the incidence of nausea between patients less than or greater than 65 years of age.5 However, the current study did find that patients with a higher baseline BMI were less likely to discontinue from the study because of nausea, which has not been reported previously. There is a possibility that the discontinuation rates may reflect patient self-perception of body image and the subsequent reaction to nausea. For example, patients with a high BMI may be more accepting of any potential weight loss associated with nausea and therefore will not discontinue from the treatment. However, these findings are a result of a posthoc subgroup analysis and, therefore, interpretation of the results is limited and should be verified by additional analyses and prospective studies.
Duloxetine was effective (based on change in HAMD17 score) at the end of the study (after 8 weeks of treatment), regardless of the dosing strategy patients received during the first week of treatment, which is in agreement with effectiveness findings from other studies using an initial dosing strategy.8,10 In addition, the rates of response and remission that were observed (48.9% and 37.0%, respectively) are somewhat similar to studies conducted in East Asian (60.5% and 49.2%; 60 mg QD for 8 weeks)3 and Caucasian (65% and 43%; 60 mg QD for 9 weeks)6 populations. These findings should reassure clinicians that a low initial dose, which may reduce nausea when patients are most susceptible, does not have a negative effect on the 8-week effectiveness of duloxetine.
The strength of the study was that it was randomized and conducted in a Korean-only population. Therefore, the findings are relevant to clinical practice in Korea. In addition, conducting the study in a mostly homogeneous population overcomes any differences that may occur in the response to treatment for depression or mental illness because of ethnicity.14 However, this study is limited mainly by the large number of protocol violations, which resulted from early trial difficulties and issues with trial management. Other limitations that may have biased the outcomes were that the dose and primary endpoint were not blinded, the food intake was not specified to patients, food and medication intake were not recorded by patients, the use of emetogenic medications, and the study was focused on nausea and gastrointestinal effects by using a solicited adverse events scale, which may have sensitized patients and physicians to nausea. The open-label design meant that both patients and investigators were aware of the dose regimen and that nausea was the study's primary endpoint. Expectation bias can occur in an open-label study,15 when patients may err in reporting and measuring data towards an expected outcome, in this case nausea.
In conclusion, this study suggests that Korean patients with major depressive disorder who require duloxetine treatment should be given a dose of 30 mg QD, with or without food, for the first week of treatment, followed by 60 mg QD for the course of therapy. In addition, to prevent discontinuation because of nausea, clinicians are recommended to highlight to Korean patients with a low BMI that nausea is generally mild to moderate and is likely to resolve in just over a week.
Acknowledgments
This study was sponsored by Eli Lilly and Company. In compliance with the Uniform Requirements for Manuscripts, established by the International Committee of Medical Journal Editors, the sponsor of this study did not impose any impediment, directly or indirectly, on the publication of the study's results. The authors acknowledge the independent medical writing assistance provided by Janelle Keys, PhD, and Serina Stretton, PhD, of ProScribe Medical Communications (www.proscribe.com.au), funded from an unrestricted financial grant from Eli Lilly. ProScribe's services complied with international guidelines for Good Publication Practice (GPP2). The authors also acknowledge Gavan Harrison (Eli Lilly) for his editorial and administrative assistance and George Zhao (Eli Lilly) for his assistance with the statistical analysis. The authors would like to thank all patients who participated in the clinical trial.
Notes
RW, JR, and MSK are full-time employees of Eli Lilly and company. YMA has received honoraria from Eli Lilly and Company for consultation and participation in advisory boards. MSL and SC have no relevant financial interests to declare.