Changes in Cerebral Cortex and Limbic Brain Functions after Short-Term Paroxetine Treatment in Panic Disorder: An [18F]FDG-PET Pilot Study
Article information
Abstract
Objective
Panic disorder (PD) is a common and often chronic psychiatric illness, and serotonin-specific reuptake inhibitors (SSRIs) are the drugs of choice for the treatment of PD. Previous studies suggested the cerebral cortex and limbic brain structures played a major role in the development of PD, but the therapeutic effect of SSRIs on specific brain structures remains unclear in PD. We examined the changes in PD patients' glucose metabolism using the [18F] Fluorodeoxy-glucose-positron emission tomography (FDG-PET) before and after 12 weeks of paroxetine treatment.
Methods
We assessed the brain glucose metabolism of 5 PD patients, using the [18F]FDG-PET, and treated them with paroxetine (12.5-37.5 mg/day) for 12 weeks. Then, we compared before and after treatment PET images of the patients, using voxel-based statistical analysis and a post hoc regions of interest analysis. Furthermore, we measured the patients' clinical variables, including information from the Panic Disorder Severity Scale (PDSS), Clinical Global Impression for Severity (CGI-S), and Hamilton Anxiety Rating Scale (HAMA).
Results
After 12 weeks of paroxetine treatment, the patients showed significant clinical improvement in terms of PDSS, CGI-S and HAMA scores (12.8±1.8 vs. 3.8±2.3, 4.6±0.5 vs. 2.0±1.4, and 15.2±4.0 vs. 5.0±1.2, respectively; all p values<0.05). After treatment, patients' glucose metabolism increased significantly in global brain areas: the right precentral gyrus, right middle frontal gyrus, right amygdala, right caudate body, right putamen, left middle frontal gyrus, left precentral gyrus, left insula, left parahippocampal gyrus, and left inferior frontal gyrus (All areas were significant at uncorrected p<0.001 and cluster level corrected p<0.05).
Conclusion
In these PD patients, cerebral cortex and limbic brain functions changed after short-term treatment with paroxetine. The therapeutic action of paroxetine may be related to altered glucose metabolism at both the cerebral cortex and limbic brain areas.
INTRODUCTION
Panic disorder (PD) occurs in approximately 3.5% of the general population and in up to 20% of primary care patients.1,2 This relatively common syndrome remains a chronic illness, despite the availability of effective anti-panic treatments, such as serotonin-specific reuptake inhibitors (SSRIs). While most patients show a clinical response to SSRIs, only 30-40% of them experience a cure.3 Thus, for better treatments, clinicians need more information about SSRIs' therapeutic mechanism in PD.
There have been many biological investigations into the pathophysiology of PD. Studies have implicated abnormal functioning in catecholamines (noradrenergic and dopaminergic) and serotonergic and GABAergic systems, as well as abnormal chemoreceptor reactivity, in the pathophysiology of PD.4 Medications that are thought to interact with monoamines and serotonergic systems, such as tricyclic antidepressants, monoamine oxidase inhibitors, and SSRIs are effective in treating PD, suggesting monoamine neurotransmitters potentially play a role in PD.5
There have been reports that PD patients show neural processing abnormalities in several brain regions, such as the frontal lobe, limbic system, and temporal lobe.6 Imaging investigations, especially functional neuroimaging studies, are in the highlight as research modalities for establishing the pathophysiological mechanisms of psychiatric illnesses. Among them, PET permits visualization of regional brain metabolism and neuroreceptor systems by means of a positron-labeled tracer and a quantitation model. As PET is more sensitive to post-treatment brain metabolism changes, compared to other modalities, it is preferred for clinical treatment studies in patients with PD.7
A few studies have examined PD patients' brain resting states in terms of brain glucose metabolism. Using resting state PET, Reiman et al.8 showed unmedicated PD patients had abnormal asymmetry in cerebral blood flow (left less than right) within a region of the parahippocampal gyrus, as compared to normal controls. Bisaga et al.9 reported significantly increased glucose metabolism in the left hippocampus and parahippocampal area in female PD patients.
Antidepressants, especially SSRIs, are widely used as first-line pharmacological agents for PD treatment, but little is known about the change in brain function after antidepressant treatment. This pilot study measured the changes in brain glucose metabolism, using the [18F] Fluorodeoxy-glucose-positron emission tomography (FDG-PET), in PD patients before and after 12-weeks of paroxetine treatment and examined the therapeutic effect of paroxetine on brain structures in PD.
METHODS
Participants
Five patients who met the DSM-IV criteria for current PD and had a Panic Disorder Severity Scale (PDSS) score over 7 participated in this study.10 All subjects were right-handed. The patients were recruited from the outpatient psychiatric unit of Samsung Medical Center in Seoul and diagnosed using the Structured Clinical Interview for the DSM-IV.11 The clinical evaluation included a physical examination, electrocardiogram, clinical laboratory tests including liver, kidney, and thyroid function tests, and urinalysis, to rule out serious medical illnesses. We measured the clinical severity of the PD using the PDSS, the Clinical Global Impression for Severity (CGI-S), the Hamilton Rating Scale for Depression (HAM-D),12 the Hamilton Rating Scale for Anxiety (HAM-A),13 and the Spielberger State-Trait Anxiety Inventory,14 both before and after the treatment.
Exclusion criteria for all subjects included current major medical or neurological disorders; coexisting major psychiatric illnesses, including current major depression; any anxiety disorder other than PD; lifetime history of bipolar disorder, schizophrenia, and substance use disorders; and exposure to psychotropic medications within 4 weeks of PET scanning (within 8 weeks for fluoxetine). Subjects with a HAM-D score >17 were also excluded.15 Additional exclusion criteria included a personal or first-degree family history of major psychiatric illnesses. All subjects abstained from alcohol and caffeine for 24 hours before the PET scan. The Institutional Review Board of Samsung Medical Center approved this study, and all subjects gave their informed consent before participating in the study.
Drug regimen
We treated all PD patients with paroxetine medication for 12 weeks,16 following baseline evaluations. No cognitive behavioral treatment was combined with this pharmacotherapy. The initial paroxetine dose for PD patients was 12.5 mg/day for the first week. The physician determined doses administered during treatment based on adverse events and clinical treatment responses. Two patients received adjunctive alprazolam p.r.n. during the first 3 weeks. The mean final dose of paroxetine was 27.0 mg.
PET procedures
We conducted the PET scans at the Department of Nuclear Medicine of the Samsung Medical Center, obtaining PET measurements of brain glucose metabolism at baseline and again at the end of treatment for all participants. Baseline scans took place during the week before the start of treatment, and final scans within 1 week of the last treatment visit. All PET sessions began at 8-9 : 00 AM. Using a GE Advance PET scanner (GE Medical Systems, Milwaukee, WI), we acquired the images with participants awake and rested, with their eyes closed and ears unplugged. For each scan, we administered 4.8 MBq/kg of [18F]FDG via a peripheral vein catheter and recorded emission data over 15 minutes, starting 30 minutes after the [18F]FDG injection. A customized fitted thermoplastic facemask minimized participants' head movements while allowing for accurate repositioning. We used a Hanning filter (cut-off frequency=4.5 mm) to reconstruct the PET images and displayed them in 128×128 matrix (pixel size=1.95×1.95 mm with a slice thickness of 4.25 mm). We acquired PD patients' post-treatment PET images using the same scanner and an identical protocol.
Data analysis
We analyzed the changes in the clinical psychological data between the baseline and post-treatment by means of a paired t-test and performed the statistical analyses of the PET data using statistical parametric mapping (SPM) 2 (Wellcome Department of Imaging Neuroscience, University College London, London, England), implemented in Matlab version 7.0. To determine the stereotactic coordinates of the peak differences, we used the Talairach and Tournoux17 atlas, after translating the MNI coordinates to coordinates according to the Talairach template via the Matlab function mni2tal.m (http://imaging.mrc-cbu.cam.ac.uk/imaging/MniTalairach). Before spatially normalizing the pre-treatment and post-treatment PET images to a standard FDG-PET template, we linearly transformed each pre-treatment PET image to match the post-treatment PET image. Also, we normalized all of the PET images to the standard FDG-PET template in SPM2, using a 12-parameter affine and a non-linear transformation, and smoothed them using a 14-mm, full-width, half-maximum Gaussian kernel to increase the signal to noise ratio. We normalized the count of each voxel to the total count of the brain (proportional scaling) to remove the differences in global glucose metabolism between individuals. To examine the statistical significance of the differences between pre-treatment and post-treatment, we utilized a paired t-test (statistical significances were set at uncorrected p<0.001). Finally, we calculated the adjusted mean activities of the regions of interest (ROIs), by the previously-described method, from the clusters obtained from voxel-wise t-statistics and performed a post-hoc ROI analysis to examine the correlation between the change in brain activity and the change in psychological variables.18
RESULTS
Table 1 shows the patients' demographic and baseline clinical characteristics (n=5). The patients' mean age was 42.6 years, and 2 of the participants had agoraphobia. All patients were naïve to psychotropic medications except paroxetine, and 2 had taken benzodiazepines intermittently. No PD patients experienced a panic attack during PET scanning.
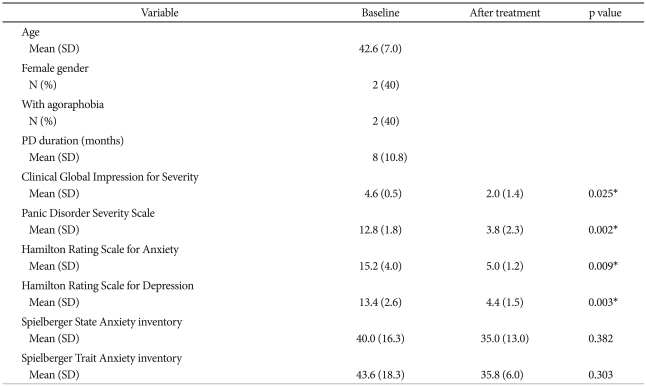
Demographic and clinical characteristics (N=5)
All patients responded to the 12 weeks of paroxetine treatment (no panic attacks during the final 4 weeks and final PDSS score <50% of baseline PDSS score). The patients showed clinical improvement in terms of the CGI-S [4.6 (0.5) vs. 2.0 (1.4); p=0.025], the PDSS [12.8 (1.8) vs. 3.8 (2.3); p=0.002)], the HAM-A [15.2 (4.0) vs. 5.0 (1.2); p=0.009)], and the HAM-D [13.4 (2.6) vs. 4.4 (1.5); p=0.003)].
After the 12 weeks of paroxetine treatment, patients showed a significantly increased [18F]FDG uptake compared to baseline in 10 brain areas: the right precentral gyrus, the right middle frontal gyrus, the right amygdala, the right caudate body, the right putamen, the left middle frontal gyrus, the left precentral gyrus, the left insula, the left parahippocampal gyrus, and the left inferior frontal gyrus (Table 2, Figure 1).
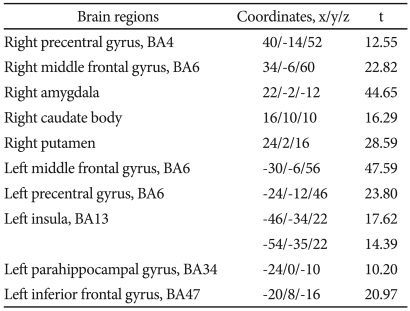
Regions where [18F] FDG uptake increased after paroxetine treatment in patients with panic disorder, by voxel-wise analysis*
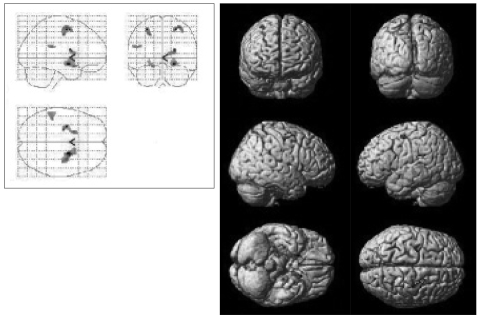
Statistical parametric map illustrating brain areas with increased activity, in panic disorder patients (N=5) after 12 weeks of paroxetine treatment.
DISCUSSION
Previous studies of anxiety disorder emphasized the role of the limbic brain and the cerebral cortex regions.19-22 In line with our results, Boshuisen et al.23 reported PD patients had hypoactivity in the precentral gyrus, the inferior frontal gyrus, and the anterior insula, both during anticipatory anxiety and at rest after pentagastrin challenge. Unfortunately, our study did not include a control group, and we did not know which brain structures of the patients were abnormal compared with those of normal subjects. However, our results suggest a link between cerebral cortex regions and SSRI treatment in terms of brain glucose metabolism. Similar to previous studies, we could detect the functional changes after treatment in the hippocampal and parahippocampal areas or the amygdala in patients with PD. Functional or structural abnormalities of the hippocampus, parahippocampus, and amygdala are often reported in PD.6 Our findings suggest that cerebral cortex regions, as well as limbic brain areas including the amygdala, hippocampus, and parahippocampal gyrus, may play a more important role in PD treatment. This study is the first to examine the effect of a single SSRI on brain metabolism in PD patients and suggests that paroxetine treatment may work its therapeutic action by increasing the activities of not only the limbic brain but also cerebral cortex regions, including the prefrontal cortex.
Gorman et al.6 suggested a possible mechanism for the two different main treatment modalities, SSRIs and cognitive behavior therapy (CBT), in PD patients. The hypothesis is that medications, particularly those that influence the serotonin system, desensitize the fear network, from the level of the amygdala through its projections into the hypothalamus and the brainstem.24 Effective psychosocial treatments may also reduce contextual fear and cognitive misattributions at the level of the prefrontal cortex and hippocampus.25 Prasko et al.26 examined the effects of SSRIs, as well as CBT, on PD patients in terms of basal glucose metabolism, using [18F]FDG-PET. They reported SSRIs cause metabolic changes at the cortical level, such as decreases in the inferior temporal gyrus and the right hemisphere's superior and inferior frontal gyri, increases in the inferior frontal gyrus and middle temporal gyrus, and (increases in) insulin in the left hemisphere. They did not detect changes in [18F]FDG uptake in the limbic regions (hippocampus, parahippocampal gyrus, and amygdala). However, their study had some limitations, such as using a priori hypothesized ROIs and using different medications on each patient.
Icreased metabolism in the right amygdala and left parahippocampal gyrus after paroxetine treatment seems paradoxical, because the amygdala and parahippocampal gyrus important structures in fear acquisition and consolidation. Many studies have shown that acutely increasing 5-HT levels, either globally or regionally, in the brain of an experimental animal increases fear and avoidance.27 In some studies, long-term administration of 5-HT precursors or agonists appear to maintain this fear increase,28 although some contradictory data exist. This stimulating effect of serotonin seems to be related to increased metabolism in the amygdala and the parahippocampal gyrus.29 However, the stimulating effect of serotonin in PD patients is usually temporary, and SSRIs are effective in reducing panic symptoms. Thus, in PD, paroxetine's effect may result from increased frontal cortex activity, whereas the paroxetine's role in the amygdala and the parahippocampal gyrus is still in debate.
Our study had several limitations. First, as this was a pilot study, our sample size was too small. Examining the exact pathophysiology of PD woud require a larger patient sample size, with a control group. Second, this study's design was semi-naturalistic, and the medication dosages were flexible, according to the physician's decisions. Finally, the relatively short pharmacotherapy duration may be a potential limitation in assessing our results.
In conclusion, PD patients' cerebral cortex and limbic brain functions changed after short-term treatment with paroxetine. Paroxetine's therapeutic action may be related to altered glucose metabolism in both the cerebral cortex and limbic brain areas.
Acknowledgments
This study was supported by the Choi Shin Hae Research Fund of Korean Neuropsychiatry Research Foundation in 2007.