INTRODUCTION
Schizophrenia and bipolar disorder are devastating mental illnesses that can lead to deterioration in the social and occupational functioning of affected individuals [1,2] with a major cost to society [3,4]. A wide range of studies suggest a genetic component to the inheritance of both of these psychotic disorders [5]. Although bipolar disorder and schizophrenia were historically considered distinct illnesses, a slew of recent studies suggest that these disorders are genetically overlapping [6-12]. Previous studies conducted in the Costa Rican population have shown evidence of linkage disequilibrium between markers within the 18q21 region and both of these psychiatric phenotypes, i.e. severe bipolar disorder and schizophrenia [12,13] Outside the Central Valley of Costa Rica (CVCR), three separate linkage studies have previously pointed to a gene predisposition locus in this general region for schizophrenia (SC) [14], bipolar disorder (BP) [15], and both BP and SC [16]. Further analyses revealed that psychotic symptomatology (hallucinations, delusions, disorganized thought, disorganized behavior) was the relevant phenotype associated with the 18q21 region, as both persons with and without a history of mania show evidence of linkage disequilibrium in this region [12]. Psychotic symptoms are a core feature of both schizophrenia and schizoaffective disorders and are often seen in acute phases of bipolar disorder.
Many complex genetic effects including epigenetic effects presumably operate via mechanisms in gene-uterine interaction. A popular design for the investigation of such effects, including effects of parent-of-origin (imprinting), maternal genotype, and maternal-fetal genotype interactions, is to collect DNA from the affected offspring and their mothers (case/mother duos) and to compare it with an appropriate control sample. An alternative design uses data from cases and both parents (case/parent trios), but does not require controls.
The current era of genome-wide association studies has popularized the case/control design for the detection of genetic variants predisposing to complex diseases. However, as recently indicated [17], associations detected in a case/control study can arise from genetic effects as well as alternative mechanisms that are statistically confounded with case genotype effects, such as maternal genotype effects, maternal-fetal interactions, and parent-of-origin effects. A variety of diseases, particularly those related to pregnancy outcomes or complications in utero, have been hypothesized to operate via such mechanisms. For example, both maternal and fetal genes, either individually or in combination, have been implicated in the risk of pre-eclampsia [18-20], low birthweight [21,22], spina bifida [23], and schizophrenia [24]. With data collected only on cases and controls, these different types of effects will be indistinguishable. For example, a strong maternal genotype effect may present the same pattern of risks as a weak offspring (case) genotype effect, since cases and mothers of cases share an allele. However, with data from families rather than case/control data, specifically with genotype data for cases plus their mothers and/or fathers, it may be possible to distinguish between these different mechanisms [25-31].
A popular design for the investigation of maternal effects and maternal-fetal interactions operating perhaps via geneuterine interactions is to collect DNA from offspring and their first-degree relatives [27]. A comparison of the genotype relative risks in cases displaying some disease of interest vs. controls compared to the relative risks in mothers of cases vs. mothers of controls can allow investigation of the merits of different competing underlying models. For example, unusual risk patterns were found when analyzing children affected with clinical signs of congenital toxoplasmosis vs. controls, compared to when analyzing mothers of affected children vs. mothers of controls, a result that was interpreted as indicating the presence of a maternal genotype and/or imprinting effect [32].
More formally, with genotype data from ‘‘duos’’ consisting of offspring together with their mothers, one could fit models that incorporate the effects of offspring genotype, maternal genotype, maternal-fetal interactions, and imprinting [27,33-35].
A number of authors have considered the alternative approach of using case/parent trios for estimating such effects [25,26,28-30,33]. Case/parent trios are often used in genetic association studies because of the robustness they can provide to population stratification, via the use of family-based tests that examine the transmission of high-risk alleles from parents to the affected offspring [36]. However, the case/control design has recently obtained greater popularity owing to the larger sample size [37] and development of alternative methods to deal with population stratification [38-40]. With case/parent trios, we can test for association and estimate genotype and haplotype relative risks using conditional logistic regression [41-43] or log-linear modeling [44].
More complex effects such as maternal genotype effects, maternal-fetal interactions, and parent-of-origin effects may be estimated through an extension of the conditional logistic regression approach [25] or through log-linear modeling [28-30,45]. One of the merits of the case/parent trio design is the fact that it does not require control data: essentially, the untransmitted parental alleles or genotypes are used as ‘‘controls’’ for the transmitted alleles or genotypes.
We investigate the effects of estimating maternal, imprinting, and interaction effects using multimodal modeling in parents and their offspring with schizophrenia in a Korean population.
METHODS
Sample collection
All subjects were recruited in accordance with the principles of the Declaration of Helsinki and with approval from the Institutional Review Boards of Pusan National University Hospital in South Korea (IRB No. 0908-003-001), and gave informed consent. Probands were recruited independently from psychiatric hospitals and clinics in the Kyungnam Province, South Korea. The Korean population in Kyungnam Province is ideal for conducting family-based association studies as it is a “founder” population [46] with characteristics that enhance the likelihood of finding linkage disequilibrium between disease-causing genes and specific markers [13,47]. All probands showed disease onset by age 40, a history of at least one psychiatric hospitalization, a discharge diagnosis of schizophrenia, Korean surnames, and a current residence in the region. These probands all had grandparents who had been born in the region, as documented by genealogical analyses. Parents and first-degree relatives of the probands were also recruited wherever possible, to permit determination of genetic phase and to allow for family-based linkage disequilibrium analyses; if additional relatives with psychotic disorders were detected, efforts were made to recruit these relatives as well. We used linkage disequilibrium design for pedigree recruiting. We recruited probands with their parents and siblings whenever possible. In our study, first degree relatives include siblings, parents, or grandparents. The EMIM program adopts possible probands and mothers or parents of their first-degree relatives for analyses [12].
As in previous linkage studies on bipolar disorder in a Hispanic population [46,48], each subject was interviewed by a trained psychiatrist who was blinded to the previous history of the subject, using the Korean version of the Diagnostic Interview for Genetic Studies (DIGS) [49,50]. Medical records were also abstracted. An interview with a close relative, using a Korean version of the Family Interview for Genetic Studies (FIGS) [50,51] was also completed for each subject. Twenty seven probands and their available family members were analyzed in the current study. All affected subjects within each family were diagnosed using a best estimate diagnostic process [12]. The process arrives at a lifetime consensus diagnosis or diagnosis using the DSM-IV. The number of subjects and families analyzed in this study was as follows: 59 subjects from 27 families, with an average of 2.2 subjects genotyped per family. Of these 59 subjects, all probands had Axis I disorders according to the consensus DSM-IV diagnoses with schizophrenia. The remaining 32 family members were classified as phenotype unknown for the purpose of statistical association analyses.
In total, 27 subjects had a history of psychosis, operationalized in this study as the presence of at least one of the following at some point during their lifetime: hallucinations, delusions, grossly disorganized thought processes, or grossly disorganized behavior. This definition of psychosis corresponds to four of the five symptoms and signs listed under Criteria A of the DSM-IV definition of schizophrenia. We did not consider a history of negative symptoms, which are also listed under Criteria A of the DSM-IV definition of schizophrenia, as sufficient for a diagnosis of psychosis. For each subject, the best estimators also diagnosed whether or not manic syndromes or episodes had been present during the course of the disorder (Table 1).
Genotyping
DNA from 59 people, 27 of which were affected with a psychotic disorder, was first genotyped using 20 single nucleotide polymorphisms from D18S450 to D18S484. Given the parallel finding from previous studies that the ME2 gene was a potential candidate gene in this region, we genotyped the entire sample using twenty SNPs covering the region spanning the ME2 gene (which lies between D18S473 and D18S474), at an average inter-marker distance of 71 kb (Table 2). Standard PCR was performed using the ABI 877 automated thermocycler or the PE 9700 PCR instrument. Amplified fragments were analyzed on the 3100 Genetic Analyzer for microsatellites and on the 7900HT Sequence Detection System for SNPs. Genotypes were assigned using GeneScan, Genotyper, and SDS software. Each genotype was scored separately by two individuals, who were blinded to the diagnosis of the subjects. Genotype scores were compared using a software program implemented in Microsoft Excel, discrepancies were discussed with a review of the original gels, and final genotypes were agreed upon. Genotypes were checked for violations of Mendelian inheritance using the PEdigree Database SYStem (PEDSYS) program, INFERence (INFER). One family was discarded from the statistical analyses due to recurrent Mendelian discrepancies. We used PBAT-tools for power calculations of binary traits in family-based studies.
Statistical analysis
EMIM tests with the candidate genes
In the fine mapping stage where 17 single nucleotide polymorphism markers were tested, Estimation of Maternal, Imprinting and interaction effects using Multinomial modelling (EMIM) was performed for the phenotypes of “schizophrenia”. All other subjects were classified as unknown for purposes of EMIM analyses. For testing of association with the candidate genes, we performed EMIM analyses with seventeen individual SNPs within the candidate genes that were determined to be in strong linkage disequilibrium with the ME2 SNPs, using the phenotypes of schizophrenia. EMIM is a FORTRAN 77 program that compiles under Linux or Windows (DOS) using a suitable compiler such as g77, gfortran, fort77, and f77. Compiling on any other system has not been attempted. However, there is no reason why the program should not compile provided an appropriate FORTRAN 77 compiler is available. Use of the command line interface (i.e. DOS command prompt) is required to run EMIM under Windows [54].
RESULTS
Linkage disequilibrium analyses
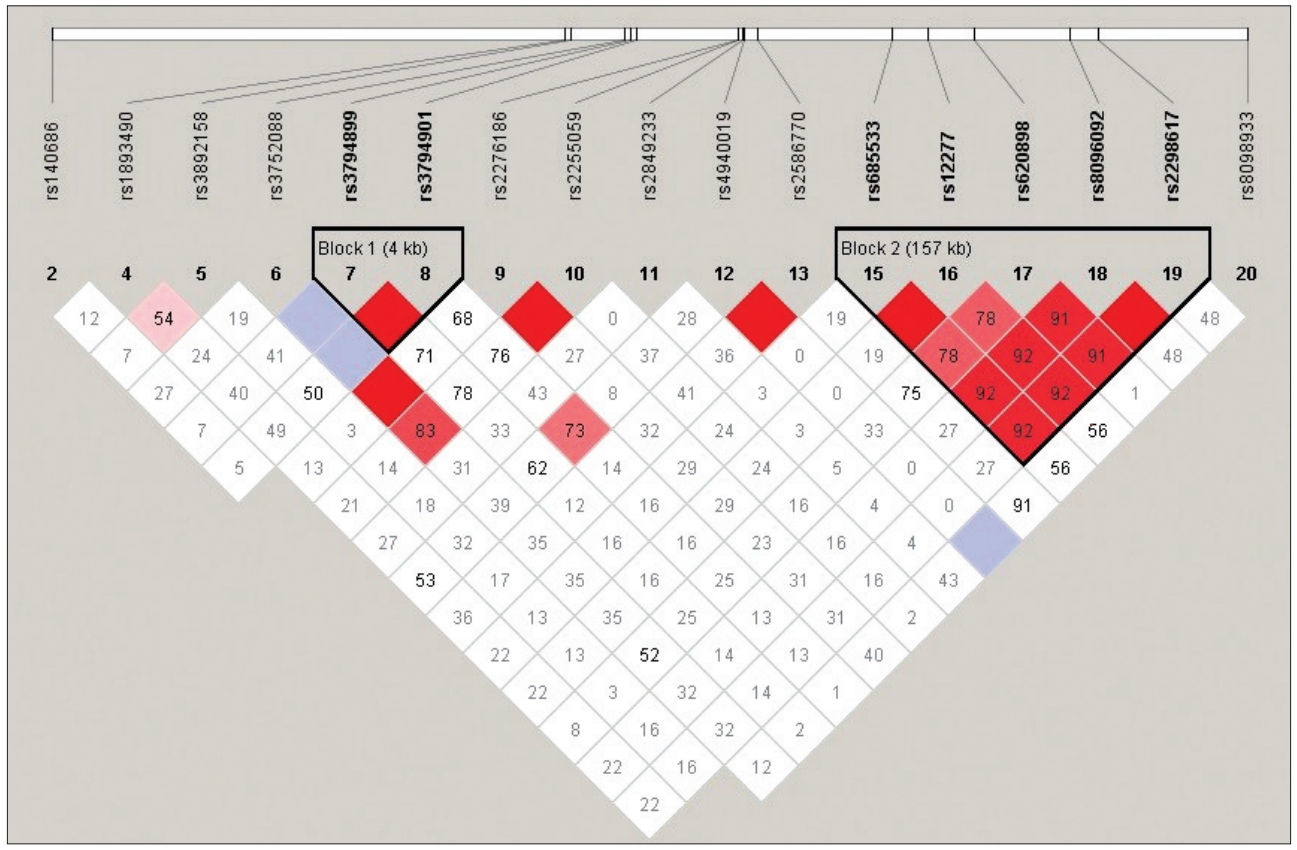
Linkage disequilibrium analyses of all microsatellites and SNPs in this region, using the Arlequin software package, revealed at least some evidence of linkage disequilibrium (p<0.05) across the entire region from D18S450 to D18S484. Regions with strong linkage disequilibrium were less extensive as shown in Figure 1. Six SNPs selected in this study were in strong LD with each other. Since they are in LD, they do not actually provide any additional information on the association of genes to the phenotype of psychosis. Therefore, we excluded overlapping SNPs in the subsequent association and haplotype analyses.
Single nucleotide polymorphism EMIM analyses
Table 2 displays the results of EMIM analyses for individual single nucleotide polymorphism markers in the 18q21 region. Individual single nucleotide polymorphism markers showing an association to schizophrenia with global p values below 0.05 were contiguous markers, both at the same genetic distance. These are nominal p values. Utilizing a standard Bonferroni correction for multiple testing would require a nominal p value below 0.0025 for a significant evidence of association. Such a correction may be overly stringent if markers are in linkage disequilibrium with each other.
The ME2 gene lies between D18S473 and D18S474. Given the findings of under-expression of this gene in previous studies, we focused our subsequent analyses on the candidate genes around this gene. We genotyped and analyzed the results for 20 SNPs placed between D18S473 and D18S474, to determine the SNPs that were in strong linkage disequilibrium with the ME2 gene. Two of the SNPs were within the MBD1 gene, six within the MAPK4 gene, five within the MRO gene, three within the ME2 gene, one within the ELAC1 gene, two within the SMAD4 gene, and the other one was on the MEX3C gene. Analysis of linkage disequilibrium between these SNPs revealed that two SNPS located within the MAPK4 gene, plus SNPs within the ME2, ELAC1, and SMAD4 genes were in strong linkage disequilibrium with each other (D’>0.60) (Figure 1). In order to test the hypothesis that candidate genes will be associated with the phenotypes of interest, subsequent analyses were performed for these twenty SNPs. The twenty SNP markers were first analyzed individually. Using Bonferroni correction for testing these twenty SNPs and one phenotype would require a global P value below 0.0025 for significant evidence of association. No SNP showed significant evidence of association to the schizophrenia phenotype.
Of the 20 SNPs analyzed, a significant SNP (rs 2276186) was suggested in EMIM analysis for child genetic effects (p=0.0225438044) and child genetic effects allowing for maternal genetic effects (p=0.0209453210) with very stringent multiple comparison Bonferroni’s correction. Additionally, analysis results for maternal genetic effects and maternal genetic effects allowing for child genetic effects was presented.
DISCUSSION
The present findings provide convergent evidence of fine EMIM mapping of a chromosomal locus associated with schizophrenia suggesting that MRO is a candidate gene causing a spectrum of the schizophrenia phenotype. This gene is specifically transcribed in males before and after differentiation of the testis, and the encoded protein might play an important role in mammalian sex determination. Multiple transcript variants encoding different isoforms have been found for this gene.
Previous studies conducted in the Central Valley of Costa Rica (CVCR) have independently shown evidence of linkage disequilibrium between these phenotypes and the 18q21 region [12,13], spanning a 2.41 cM region (2.7 Mb) from D18S450 to D18S474. Outside of the CVCR, three separate linkage studies have previously pointed to a gene predisposition locus in this general region for SC [14], BP [15], or both BP and SC [16]. A study obtained evidence of linkage in a combined sample of SC and BP at marker D18S472, which is at approximately the same position as marker D18S474 in the present study [16].
The present study provided fine EMIM analyses for the 18q21 locus in the Korean population. Previous analyses first narrowed the area of strongest association with the psychosis phenotype to the region from 45.74 Mb (D18S473) to 46.94 Mb (D18S474). There are 11 known genes that are located within this 1.20 Mb region: MYO5B (45.60 Mb-45.98 Mb), FLJ32743 (46.01 Mb-46.05 Mb), MBD1 (46.05 Mb-46.06 Mb), CXXC1 (46.06 Mb-46.07 Mb), C18orf24 (46.16 Mb-46.17 Mb), LOC390853 (46.24 Mb-46.34 Mb), MAPK4 (46.44 Mb-46.51 Mb), MRO (46.58 Mb-46.60 Mb), ME2 (46.66 Mb-46.73 Mb), ELAC1 (46.75 Mb-46.77 Mb), and SMAD4 (46.81 Mb-46.86 Mb). Although the SNP association analyses presented here focuses on the MRO gene, three of the genes in this region (MBD1, ME2, and SMAD4) are of special interest due to their known functions and/or their relationship of association with neuropsychiatric disorders. MBD1 gene is known to be associated with Rett disorder and autistic disorders [55,56]. ME2 gene has shown evidence of association with idiopathic generalized epilepsy, and the strongest association is seen with variants in the promoter region of this gene [57]. The SMAD4 gene contains a single SNP showing the strong evidence of association with psychosis in the previous study and is therefore an interesting potential candidate gene for schizophrenia and bipolar disorder. It is a member of the SMAD family of transcription factors that are activated by transforming growth factor-β receptors [58], and is known to be involved in neuronal proliferation and differentiation [59]. Genotyping evidence in the previous study shows direct association of haplotypes containing SNPs within the MAPK4 gene and the schizophrenia phenotype. An individual SNP that is physically inside the MAPK4 gene, but in nominal value, also showed significant evidence of association with the phenotype of psychosis. Based on genotype analyses alone, the possibility remains that a gene variant or mutation within either of the genes in linkage disequilibrium with the ME2 gene (SMAD4 and ELAC1) might be responsible for the association detected in this study [60,61]. Further studies including sequencing for possible novel mutations in the candidate genes will be necessary in these and other samples from Korean pedigrees to more definitively assess whether candidate genes in 18q21 contain specific variants that directly cause the spectrum of psychotic illnesses seen in the current sample. Further analyses of the region spanning D18S450 to D18S484 will be required to test other genes for evidence of association with schizophrenia.
One limitation of the current study is the relatively low statistical evidence of association, and therefore, this finding should be confirmed in a larger sample from the Korean or another population sample. Linkage analyses have obvious strength in localizing the susceptibility genes of common, complex diseases such as schizophrenia. Traditionally, linkage analyses have suggested broad candidate regions for further fine mapping analyses and linkage disequilibrium analyses have mapped finely in suggestive candidate regions. Occasionally, both analyses have been used interchangeably for possible family designs that could be used regardless of the advantages. We have used linkage disequilibrium analyses for fine mapping studies of chromosome 18q21 based on possible pedigree types because restrictive pedigree types such as affected sibpairs will be required for linkage analyses. The study has the limitation of small power that is insufficient to detect an association of the modest effect sizes expected in a small sample size of 27 probands and 32 relatives in our study. However, despite this limitation, this small scale study could contribute preliminary data to genome-wide association studies (GWAS) in general and particularly to the psychiatric GWAS consortium to advance the knowledge of genetic linkages.
Another limitation is that the individual SNP showing the strongest EMIM association with schizophrenia is technically within the MRO gene and not the ME2 gene, although they are positioned finely in chromosome 18q21. Finally, denser genotyping, sequencing, and functional studies will ultimately be needed to confirm whether the MRO gene, or another gene in tight linkage disequilibrium with the candidate gene (i.e. ME2), contains a causal variant underlying the pathogenesis of psychotic spectrum disorders in the Korean and other populations. This study is just a random selection of SNPs for genotyping. We plan to have a denser genotype map or to reduce the area of the chromosome to be covered in our upcoming studies. In addition, a comprehensive study on the LD structure should be conducted before choosing the SNPs to be genotyped.
We have found few previous studies that have used EMIM analysis or reported MRO as a candidate gene in schizophrenia subjects. To our knowledge, this is the first trial epigenetic study for evaluating the effect of single nucleotide polymorphisms on gene-uterine interaction in schizophrenia. MRO (maestro) gene is specifically transcribed in males before and after differentiation of the testis, and the encoded protein may play an important role in a mammalian sex determination. Multiple transcript variants encoding different isoforms have been found for this gene. Although there are few studies on the association of this gene with schizophrenia, some studies have found that there could be some association of testicular tumors with psychotic symptoms [62-78].
S-acylation (also known as palmitoylation) is a major posttranslational protein modification in all eukaryotic cells, involving the attachment of fatty acids onto cysteine residues. A variety of structural and signaling proteins are modified in this manner, affecting their stability, membrane association, and intracellular targeting. The enzymes that mediate S-acylation are encoded by genes belonging to the large (>20 genes) ZDHHC family. The importance of these enzymes for normal physiological function is highlighted by their links to a diverse range of disease states including neurological disorders such as Huntington’s disease, schizophrenia and intellectual disability, and diabetes and cancer. A recent study by Yeste-Velasco et al published in the Journal of Pathology highlights a novel tumor suppressor function for the ZDHHC family. Expression of ZDHHC14 is decreased in testicular germ cell tumors, prostate cancer, and a variety of other cancer types. This important finding further emphasizes the emerging clinical significance of the ZDHHC family of S-acylation enzymes [79]. A recent report indicates that gonadectomy increases neurogenesis in the male adolescent rhesus macaque hippocampus suggesting that testicular development could influence neuropsychiatric symptoms by mediating the subcortical brain structures [80]. Moreover, in a large Danish cohort of schizophrenic patients, the overall incidence of cancer was reduced particularly in males. The reduced cancer incidence was particularly observed for genital cancers, particularly testicular cancer, suggesting a hypothesis that antipsychotic medications might decrease the occurrence of these neoplasms [81].
Alternative methodologies in genetic research are required with many methodological limitations in approaches evaluating the correlations between genotyping and phenotyping. Epigenetics and gene-environment interactions are represented with underlying statistical genetics. Our results are a pilot study for epigenetic studies on mental disorders and help to understand and use the EMIM statistical genetics analysis program, although with many limitations including small pedigree numbers.